
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会
中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:党双锁
执行编辑:郑素军,汤珊,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、 主编致辞
婴儿肝衰竭综合征I型(infantile liver failure syndrome type 1, ILFS1)是一种由胞质亮氨酰–tRNA合成酶基因(leucyl-tRNAsynthetase gene, LARS)突变导致的常染色体隐性遗传病。LARS基因定位于染色体5q32, 含有32个外显子, 转录子全长4766 bp,编码由1 176个氨基酸组成并位于胞质的亮氨酰-t RNA合成酶 (leucyl-t RNA synthetase, Leu RS)。截止2018年1月,国际文献总共报道LARS基因的四个突变位点,国内报道1例,检测到两个新的突变位点。
ILFS1的临床表现常涉及多个系统, 患儿通常在1岁以内表现为低出生体重(早期发育不良)、肝功能异常不全、低蛋白血症、贫血、癫痫发作,约有90%的患儿存在生长发育迟缓;当患儿出现发热性疾病时,常引起的脑病发作(80%)及间歇性肝功能障碍。此外,部分患者可能出现肝衰竭、惊厥和脑病等严重表现,甚至死亡。ILFS1患儿的肝脏病理显示显著的脂肪肝和纤维化。
本期月报报告一例以进行性黄疸加重为主要临床表现的ILFS1病例,本例患儿主要表现为低出生体重、肝功能异常、贫血、凝血功能异常、轻度低蛋白血症等,血串联质谱代谢筛查提示多种氨基酸含量增高,提示代谢紊乱,结合患儿多系统损害,高度怀疑遗传代谢病,通过二代测序分析到 LARS 基因有2个杂合突变,分别来自于父亲和母亲,该突变临床表型为小儿肝功衰竭综合征I型。通过对该病例的分析,期望临床医生提高对ILFS1的认识,对于小儿肝功能损害者,除了常见感染、药物、毒物等因素外,需警惕遗产代谢性疾病,积极运用分子诊断技术,及早明确病因。

党双锁:医学博士,主任医师,研究员,教授,博士研究生导师
西安交通大学病原与感染性疾病教研室主任
西安交通大学第二附属医院感染肝病科主任
中国医师协会整合感染与防控专业委员会副主任委员
中华医学会感染病分会常委
中国医药质量管理协会转化医学分会常委
中华医学会肝病学分会遗传代谢性肝病协作组委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
o 由新的LARS1突变引起的1型婴儿肝衰竭综合征,新生儿出现致死性肝细胞损伤和骨骼肌发育不良的严重病程(Am J Med Genet A,2021, IF=2.802; Q3区)

o 亮氨酰-tRNA合成酶缺乏系统性地诱导斑马鱼过度自噬(Sci Rep,2021, IF=4.379; Q1区
o 3'tsRNAs氨基酰化对核糖体生物合成的影响(PLoS Genet,2021, IF=5.917; Q1区)
o 1型遗传性酪氨酸血症的诊断和治疗挑战(Front Pediatr,2021, IF=3.418; Q1区)
o 胆汁淤积性肝病啮齿动物模型:转化研究的实用指南(Liver Int,2021, IF=5.828; Q2区)
o NBAS和P4HB变异导致的复杂表型(发热依赖性复发性急性肝功能衰竭和成骨不全)的特征(Mol Genet Metab,2021, IF=4.797; Q2区)
o 神经母细胞瘤扩增序列(NBAS)基因突变致小儿复发性急性肝功能衰竭1例(Front Pediatr,2021, IF=3.418; Q1区)
o 1例NBAS突变患者行肝移植治疗小儿复发性急性肝功能衰竭(Pediatr Transplant,2021, IF=1.502; Q4区)
o 基于GC-MS尿液代谢组学的随机森林同时检测多种遗传代谢性疾病(Talanta,2021, IF=6.057; Q1区)
o 通过多色探针熔解曲线分析技术快速对Citrin缺乏症的遗传诊断(Front Pediatr,2021, IF=3.418; Q1区)
三、临床资讯
1 幻灯分享
1.1标题及链接:婴儿肝衰竭综合征1型1例
1.2供稿专家简介
党双锁:医学博士,主任医师,研究员,教授,博士研究生导师
西安交通大学病原与感染性疾病教研室主任
西安交通大学第二附属医院感染肝病科主任
中国医师协会整合感染与防控专业委员会副主任委员
中华医学会感染病分会常委
中国医药质量管理协会转化医学分会常委
中华医学会肝病学分会遗传代谢性肝病协作组委员
1.3 病例分享
患儿男,5月7天,1个月前出现黄疸,因呼吸道感染黄疸加重就诊于当地医院,查CMV-IgM弱阳性,抗病毒治疗8天减轻黄染明显减轻。1天前出现发热,黄疸再次加重转诊来我院。
个人史:1胎1产,孕40+2周,因“羊水少”行剖宫产,否认出生窒息史,出生体重1.51kg,母乳喂养;4月会抬头,现不会翻身,追光追物循声可,活动量正常。
既往史及家族史:出生时因足月小样儿、新生儿低血糖留院治疗10天;否认传染病接触史;否认外伤、手术史,血液制品输注史;否认食物、药物过敏史;按程序预防接种史;家族史无异常。
入院查体:体重:4.5kg,发育较差,营养欠佳,中度贫血貌,呼吸稍促,全身皮肤重度黄染,无皮疹及出血点, 双侧呼吸音粗,可闻及细小湿性罗音。心律齐,心音有力,心前区可闻及2/6级收缩期杂音。腹壁静脉无曲张,腹稍膨隆,肝肋下可触及100px,剑下可触及50px,质韧缘锐,表面光滑,脾肋下未触及。
入院后急诊检查:肝功:TBIL 302.0umol/L,DBIL 180.5umol/L,IBIL 121.5umol/L,TP 41.2g/L,ALB 30.4g/L,ALT 388U/L,AST 1620U/L,ALP 1019U/L,GTT 77U/L;血常规:Hb 80g/L ;血氨 63.3umol/L,血乳酸4.0mmol/L,血气分析大致正常;凝血六项:PT 32.80sec,PTA 23.0% ,INR 3.20,APTT 82.10sec,Fib 1.06g/L,TT 23.40sec,补充维生素K1 4小时后复查改善不明显,给予输注新鲜血浆;腹部B超:肝右叶稍强回声,余未见明显异常;腹部CT:肝胆脾胰腺及双肾CT扫描未见明显异常。
其他辅助检查:
病原学检查:HCMV-IgM抗体阳性,血尿病毒定量阴性; EB病毒核抗原及衣壳抗原IgG抗体阳性,余阴性;微小病毒B19抗体IgM阴性;
甲状腺功能正常;
AFP>20000ng/ml;
血清铁、VitB12测定、叶酸测定正常;
心脏B超:房间隔缺损(中央型)动脉导管未闭、卵圆孔未闭;
头颅:MIR未见异常;
血串联质谱遗传代谢病筛查:标本中甲硫氨酸、酪氨酸等多种氨基酸含量升高,提示代谢紊乱,各酰基肉碱未见明显异常;
尿有机酸气相质谱分析:本次检测尿液有机酸发现辛二酸、己二酸、 葵二酸、4-羟基苯乳酸和4-羟基苯丙酮酸较参考值略有 升高,疑似代谢异常。
基因检测结果:LARS基因有两个杂合突变;c.1229A>G,p.D410G;c.2074+1G>T,分别来自父亲与母亲。
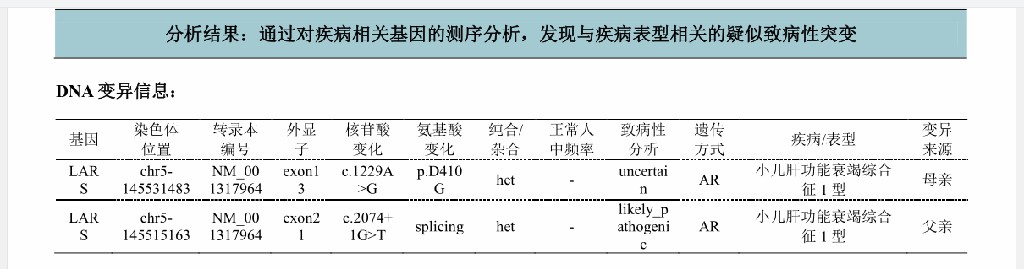
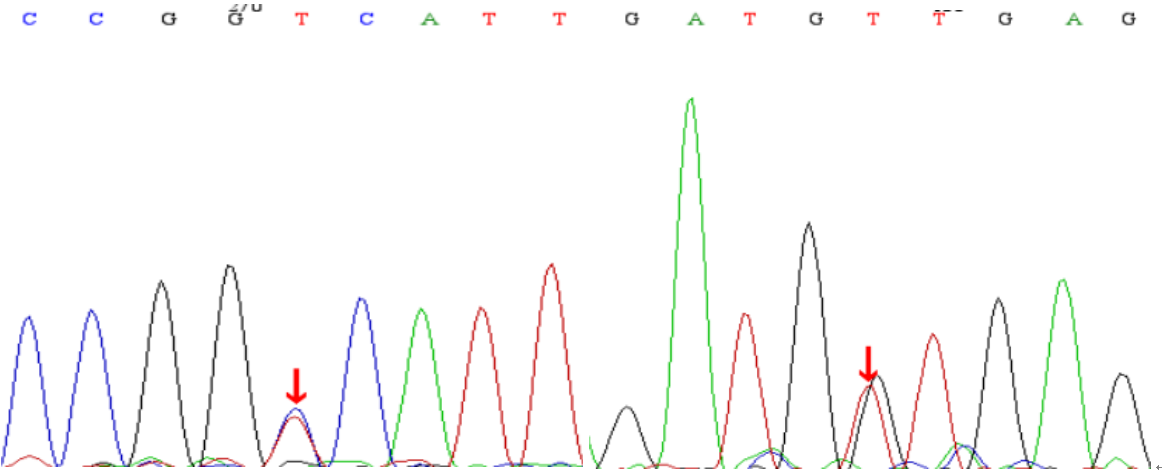
c.1229A>Gc.2074+1G>T
综上,患儿临床表现为黄疸进行性加重,查体可见生长发育差及肝脏肿大,辅助检查提示VitK1无法纠正的凝血功能异常,短期内黄疸进行性加深(TBIL 173.64→302.0umol/L),ALP 1019U/L,GTT 77U/L,存在胆汁淤积,既往无明确慢性肝病史,达到急性肝功能衰竭诊断(PT 32>20s;INR 3.20>2.0;PTA<40%)。患者足月小样儿,有新生儿低血糖病史,生长发育落后,本次入院有胆汁淤积、低蛋白血症、凝血功能异常及呼吸道感染,存在多系统损害,急性肝衰竭原因不明,结合血筛、尿筛结果,高度考虑遗传代谢病可能。研究表明,线粒体与肝衰竭的发病关系密切,肝脏受累是婴儿线粒体病变的常见临床表现,结合患儿临床表现,考虑该患儿Citrin缺陷病的可能,遂进行基因检测。线粒体全突变基因分析未发现与临床表型相关的基因突变,排除Citrin缺陷病诊断;对患儿及其父母进行基因检测,患儿基因检测结果提示LARS基因有两个杂合突变:c.1229A>G,p.D410G;c.2074+1G>T,分别来自父亲和母亲。推测两个变异位点可能为该患儿的致病变异,该突变临床表型为小儿肝功衰竭综合征I型。结合患儿临床表现,最终诊断为婴儿肝衰竭综合征1型(ILFS1)。
治疗:入院后给予拉氧头孢抗感染及干扰素雾化抗病毒治疗,丁二磺酸腺苷蛋氨酸静滴利胆退黄及多烯磷脂酰胆碱静滴保肝治疗,同时补充脂溶性维生素。患儿感染逐渐控制,肝功能及凝血功能逐渐改善,于住院治疗16天后,患儿黄疸减轻,凝血象基本正常,咳嗽消失,肺部听诊正常,好转出院。3个月后复查肝功能提示:TBIL由 302.0umol/L降至 22.4 umol/L,DBIL由 180.5umol/L降至14.0 umol/L,ALB由 30.4g/L升至39.3g/L,ALT由 388U/L降至102 U/L,AST由 1620U/L降至 108 U/L,ALP由 1019U/L降至666 U/L。9个月后再次因发热于当地医院查肝功转氨酶、胆汁酸等显著上升,白蛋白结果缺失,转来我院,入院时体温已呈下降趋势,复查肝功好转,白蛋白低,经治疗后出院前各项指标明显好转。目前随访患儿肝功能正常,凝血功能正常,无贫血,无低蛋白血症,体重及身高较正常同龄儿略迟缓,语言运动能力同正常同龄儿。
四、联系方式
o 投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
o 联系电话:010-63291007
o 联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间