
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:乔平云
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
线粒体3-羟基3-甲基戊二酰辅酶A合成酶(Mitochondrial 3-hydroxy-3-methylglutaryl-CoA(HMG-CoA)synthase,mHS)缺乏症是由HMGCS2基因突变引起的常染色体隐性疾病。mHS在酮体合成过程中催化乙酰辅酶A和乙酰乙酰辅酶A生成HMG-CoA。在长期禁食期间及碳水化合物缺乏时,酮体为大脑、心脏、肾脏提供重要的能量供应。HMGCS2致病性基因突变导致mHS缺乏,可导致禁食状态下酮体生成障碍及机体脂肪利用异常,出现低血糖、低酮体、游离脂肪酸增加、蛋白分解增加,引起代谢性酸中毒、高血氨等代谢紊乱,导致脑损伤、心肌损伤,严重时可发生昏迷及猝死,死亡率达到20%[1]。
1995年Mascaró C首次分离鉴定HMGCS2基因cDNA[2],HMGCS2基因位于1号染色体p12区域,全长2477bp,包含10个外显子,编码508个氨基酸,主要在肝脏中表达。目前已经报道的突变位点有c.1502G>A、c.1187+1G>C、c.648G>T、c.422A>T、c.559+1C>A、c.725-2A>C等,Shengnan Wu及Pengfei Zhang通过总结病例均认为mHS缺乏症中HMGCS2基因截断突变导致的临床表现更重[3]。
本病临床表现异质性大,可出现惊厥、嗜睡、呕吐、腹泻、肌张力低下、低体温、肝肿大、肌病、心肌病,甚至昏迷、呼吸暂停等。实验室检查发现代谢性酸中毒、低血糖、低酮体、血氨升高、游离脂肪酸增多、转氨酶升高,尿有机酸谱提示二羧酸尿、4-羟基-6-甲基-2-吡喃酮(4HMP)增高。尿4HMP>20μmol/mmol肌酐被认定为mHS缺乏症的特异性生化指标[4]。基因突变检测可协助明确诊断。
本病急性期的治疗包括积极纠正低血糖、代谢性酸中毒以及促进有机酸排泄。长期治疗包括避免长时间空腹,减少禁食时间,给予低脂肪、低蛋白、高碳水化合物饮食,补充左旋肉碱,保证足够的热量摄入,避免超负荷运动及感染。患儿代谢失代偿的发作将随着年龄的增长而减少,严重程度亦减轻,经及时静脉补充葡萄糖等对症治疗,患儿多数能迅速恢复。部分患儿可出现代谢危象,由能量产生严重不足引起,起病急,病情迅速进展,可危及生命。
本期月报报道一例线粒体3-羟基3-甲基戊二酰辅酶A合成酶缺乏症的女性患儿,以抽搐为主要表现,病程中还出现呕吐、嗜睡、精神差、呼吸深快等症状,化验发现低血糖、代谢性酸中毒、肝功能异常、影像学提示肝脏密度减低,考虑脂肪肝可能。该患者经过早期干预住院治疗,目前生命体征平稳,健康成长。该患儿哥哥曾于感染后出现呕吐、精神差、呻吟呼吸等症状,检查结果与妹妹相似,且具有相同的HMGCS2突变基因型,但因治疗延误于9个月时死亡。
本病是可治疗的遗传代谢性疾病,经过积极治疗预后良好,希望提高临床医生对该疾病的认识,尽早发现尽早治疗。同时,脂肪肝可能是本病的一个线索,当同时出现低血糖、代谢性酸中毒等紊乱时注意鉴别该疾病。
参考文献
1.Cotter DG, Ercal B, Huang X, Leid JM, d'Avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ, Crawford PA. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest. 2014 Dec;124(12):5175-90.
2.Mascaró C, Buesa C, Ortiz JA, Haro D, Hegardt FG. Molecular cloning and tissue expression of human mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase. Arch Biochem Biophys. 1995 Mar 10;317(2):385-90.
3.Wu S, Shen L, Chen Q, Gong C, Yang Y, Wei H, Cao B, Chen Y. Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients. Front Genet. 2022 Mar 4;12:816779.
4.Pitt JJ, Peters H, Boneh A, Yaplito-Lee J, Wieser S, Hinderhofer K, Johnson D, Zschocke J. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: urinary organic acid profiles and expanded spectrum of mutations. J Inherit Metab Dis. 2015 May;38(3):459-66.

乔平云
河南省儿童医院神经内科,副主任医师
河南省医学会抗癫痫分会癫痫共患病学组委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.病例报道:关键样本采集延迟?尿液有机酸分析还能保存一天!HMG-CoA合成酶缺乏症的新病例(Molecular genetics and metabolism reports,2024,IF=1.90; Q4区)

2.HMG-CoA裂解酶缺乏症的治疗:10例澳大利亚病例的临床和营养管理纵向数据 (Nutrients,2024,IF=5.90; Q1区)
3.曲恩汀和青霉胺对肠道铜摄取的影响:健康人代谢64 Cu PET/CT机制研究 (Hepatology,2024,IF=13.50; Q1区)
4.肝豆状核变性患者门静脉高压及其预后影响(Alimentary pharmacology & therapeutics,2024,IF=7.60; Q1区)
5.儿童和青少年肝豆状核变性的药物治疗依从性:一项来自土耳其的队列研究(Orphanet journal of rare diseases,2024,IF=3.70; Q2区)
6.进行性家族性肝内胆汁淤积2型的临床症状、生化和肝组织学特征(Orphanet journal of rare diseases,2024,IF=3.70; Q2区)
7.德国慢性酸性鞘磷脂酶缺乏症发病率和死亡率的回顾性研究(Orphanet journal of rare diseases,2024,IF=3.70; Q2区)
8.综述:肝豆状核变性:铜介导的铜死亡,铁相关的铁死毒和临床亮点,全面批判性分析更新(International journal of molecular sciences,2024,IF=5.60; Q1区)
9.综述:多囊肝的临床表现、流行病学、遗传学基础、潜在的分子靶点和当前的治疗 (Orphanet journal of rare diseases,2024,IF=3.70; Q2区)
10.共识:囊性纤维化筛查、评价和肝胆疾病管理的共识建议(Hepatology,2024,IF=13.50; Q1区)
三、临床资讯
3.1 病例分享:线粒体3-羟基3-甲基戊二酰辅酶A合成酶缺乏症1例
患儿女性,1岁4月,因“间断抽搐1月”于2023年7月至我院就诊。
现病史:患儿为母亲第2胎第2产,足月剖宫产出生,生后因呼吸费力入当地监护室之后痊愈出院。3个月竖头,6个月独坐,1岁会叫“爸爸妈妈”,1岁1月会走。1月前(2023.6.14)患儿无诱因出现抽搐,表现为双目凝视、咬牙、口唇发绀、四肢强直、意识丧失,伴有轻微腹泻、咳嗽、咳痰,不伴有发热、呕吐、皮疹等,持续约1分钟缓解,之后入睡。2天后出现发热,热峰不详。体温快速好转。2023.6.21患儿再次出现抽搐,2023.6.23患儿于睡眠中抽搐7次,每次均持续约1分钟左右自行缓解。至北京儿童医院保定医院重症监护室给予咪达唑仑泵入、鸡尾酒疗法及口服左乙拉西坦抗癫痫治疗,抽搐缓解。2023.7.7患儿至北京大学第一医院就诊停用鸡尾酒疗法,给予口服维生素B12,左卡尼汀、左乙拉西坦等治疗;2023.7.12患儿无诱因出现抽搐3次入我院,病程中有饮食差、呕吐、嗜睡精神差,检测血糖低至1.8mmol/L,给予积极补液纠正低血糖及代谢性酸中毒,左乙拉西坦加量后抽搐、精神好转,血糖恢复正常;2023.7.27再次抽搐1次,给予左乙拉西坦加量,口服左卡尼汀赖氨肌醇B12,葡醛内酯等口服,入我院仍频繁抽搐给予咪达唑仑针泵入,静点维生素B6针,口服左乙拉西坦,抽搐控制。
既往史、家族史:平素健康状况良好,有1个哥哥感染或呕吐后出现烦躁、精神差,于9个月时夭折。
体格检查:T 36.2℃,P 120次/分,R 28次/分,BW 15kg,H80cm,精神萎靡,神志清楚,全身皮肤未见牛奶咖啡斑、色素脱失斑及其他皮肤异常改变,心肺腹未见异常,四肢肌力肌张力正常,腱反射可对称引出,颅神经查体无异常,病理征、脑膜刺激征均为阴性。
实验室检查:血气分析:pH 7.304,PaCO2 36.3mmHg,HCO3-17.9mmol/L,BE-8.3,乳酸0.6mmol/L;血常规:白细胞12.92×109/L,红细胞4.24×1012/L,血红蛋白110g/L,血小板464×109/L,N 32.5%,L 55.7%,CRP<0.2mg/L;肝功能:谷丙转氨酶80.0U/L,谷草转氨酶94.0U/L,γ-谷氨酰转肽酶23.0U/L;血氨23.5umol/L;心肌酶:乳酸脱氢酶301.0U/L,羟丁酸脱氢酶234.0U/L,肌酸激酶30.7U/L,肌酸激酶同工酶17.8U/L;血糖最低1.8mmol/L。血氨基酸与肉碱谱:乙酰肉碱升高,16碳烯酰肉碱、18碳2烯酰肉碱升高,提示长链脂肪酸代谢不畅。尿有机酸检测:未见异常。
影像学检查:头颅核磁:双侧大脑半球脑沟增深,双侧脑室轻度扩张,枕大池增宽;肝脏CT:肝脏密度减低(图1)。
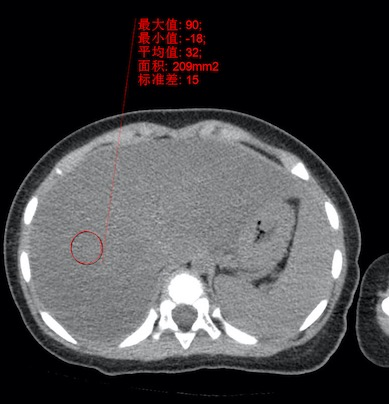
图1.肝脏CT提示肝脏密度减低(CT值32HU)
视频脑电图:异常幼儿脑电图,睡眠区额区、额中线区棘慢波发放。
全外显子基因测序结果及家系验证:患儿检测到HMGCS2基因复合杂合突变:c.220G>A(来源于父亲)、c.617G>A(来源于母亲),根据ACMG评分提示两个变异均为意义不明:c.220G>A(PP3)、c.617G>A(PM2+PM5+PP3),具体见图2、3。
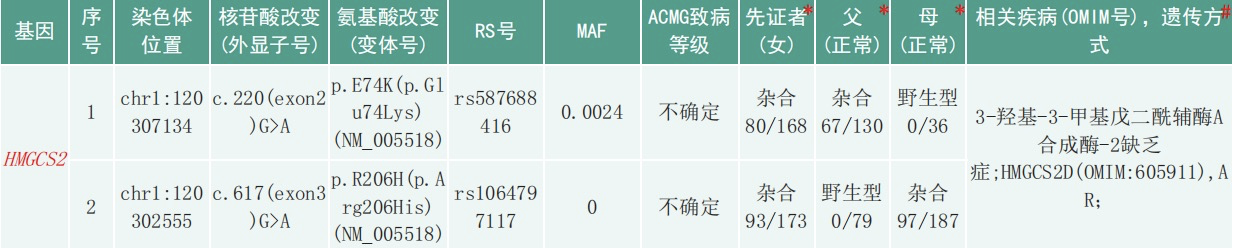
图2.患者全外显子基因测序结果
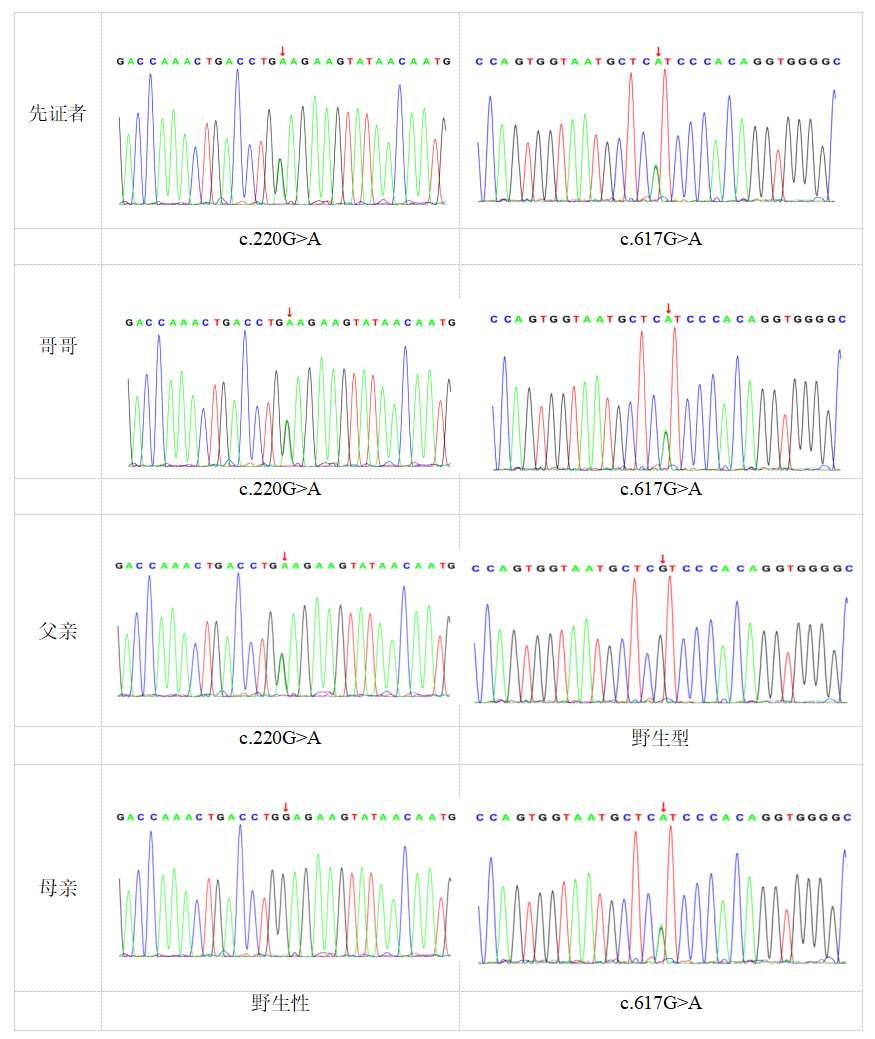
图3.患儿、患儿哥哥、患儿父亲、患儿母亲HMGCS2基因一代测序验证
最终诊断:患儿发病时有抽搐、呕吐、呼吸深快等症状,化验提示低血糖、代谢性酸中毒、肝功能异常,基因检测发现HMGCS2基因复合杂合突变,与该患儿具有相同突变基因型的哥哥于婴儿期夭折。综上,考虑诊断:线粒体3-羟基3-甲基戊二酰辅酶A合成酶缺乏症。
治疗与随访:患儿目前口服“左卡尼汀10ml qd,左乙拉西坦3ml q12h”,随访至今生长发育良好,未再发病,未再抽搐。
3.2 供稿专家简介
乔平云
河南省儿童医院神经内科,副主任医师
河南省医学会抗癫痫分会癫痫共患病学组委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间