
本文转载自“佑安肝病感染病专科医疗联盟”
主管:佑安肝病感染病专科医疗联盟(YouMe AID)
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会
指导:段钟平
总编辑:郑素军
本期责任主编:丁向春
执行编辑:白洁,孔明
本期目录:
一、主编致辞
二、学术进展
三、临床资讯
四、专业委员会名单
五、联系方式
一、主编致辞
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
遗传代谢性肝病月报主要包括以下栏目,力求“一眼抓住最新进展,短时积累临床心得!”
“学术进展”:通过文献分享、专家点评,帮您快速了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座、学会动态等信息,助您感悟遗传代谢性肝病临床诊治之奥妙!
每月一报,让遗传代谢性肝病从罕见病、疑难病变为医者眼中的熟悉病、简单病!
每月一报,助力全面提高我国遗传代谢性肝病的诊治与科研水平!
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis, PFIC)作为一组常染色体隐性遗传病,该疾病在2018年5月11日被列入国家卫生健康委员会等5部门联合制定的《第一批罕见病目录》,基因突变导致肝细胞和胆管上皮细胞上与肝内胆汁淤积相关的各种功能蛋白的生成、调控及修饰缺陷是PFIC产生的根本原因,根据致病基因不同,目前PFIC分为 6 型。临床上多见于新生儿和l岁内婴儿,以进行性的肝内胆汁淤积为主要表现,患儿常在儿童期到青春期因肝衰竭死亡,早期诊断及干预对患儿预后影响极大。在段钟平教授和郑素军教授的指导下,遗传代谢性肝病月报本期主要内容以PFIC有关的临床病例及最新文献分享为主,为临床专家今后更好的认识诊治该疾病提供更多的新的思维。在此衷心的感谢孔明教授和白洁博士的辛勤付出!愿各位专家能从本期文献分享中收获多多!

丁向春:男,主任医师,教授,博士研究生导师
宁夏医科大学总医院
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
二、学术进展
1.Pubmed收录论文汇总归类:2020年4月发表遗传代谢性肝病相关论文共37篇,按遗传代谢性肝病常见分类(比如胆红素、胆汁酸、金属、蛋白、糖等代谢异常),统计结果如下图。
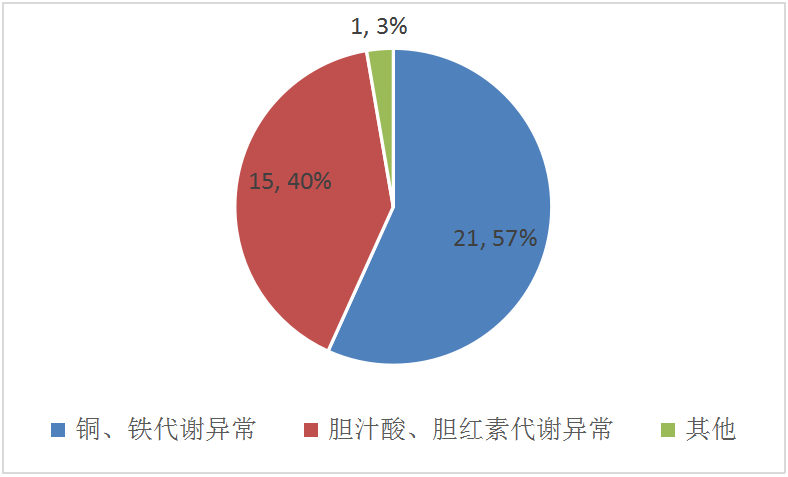
图1.2020年4月Pubmed收录遗传代谢性肝病论文及分类情况
2. PubMed重要文献速览(长按文末二维码或“阅读原文”可下载)
o 综述:威尔逊病的早期诊断与个体化治疗(Curr Neuropharmacol, 2020, IF=4.568, Q1区)

o 鼻胆管引流术可缓解进行性家族性肝内胆汁淤积症患者瘙痒症状(Eur J Pediatr, 2020, IF=2.188; Q2区)
o 长期标准化治疗可有效预防威尔逊病广泛脑损伤、减少癫痫发生(CNS Neurosci Ther, 2020, IF=3.394; Q2区)
o 杂合突变威尔逊氏病患者脑部超声有黒质、豆状核高回声表现(Neurol Sci, 2020, IF=2.484; Q3区)
o 遗传性血色素沉着症可引起高尿酸血症(Biochem J, 2020, IF=4.331; Q1区)
o 系统回顾:Crigler-Najjar综合征的疾病负担(J Gastroenterol Hepatol, 2020, IF=3.632; Q2 区)
o 利用人胚胎干细胞可构建体外威尔逊氏病模型(Cells, 2020, IF=4.829; Q1区)
o 遗传性血色素沉着症增加关节置换术几率(Semin Arthritis Rheum, 2020, IF=5.072; Q1区)
o UGT1A1的Gly71Arg和TATA启动子突变增加新生儿高胆红素血症的风险(Gene, 2020, IF=2.638; Q2区)
o 评论:威尔逊病的分子诊断挑战(J Clin Pathol, 2020, IF= 2.346; Q2区)
3.Pubmed精选文献介绍及专家简评(长按文末二维码或“阅读原文”可下载文献全文)
3.1文献一(英文):Tomoko Uehara, Mamiko Yamada, Shuichiro Umetsu, et al. Biallelic Mutations in the LSR Gene Cause a Novel Type of Infantile Intrahepatic Cholestasis. J Pediatr, 2020 Jun;221:251-254. Impact factor=3.739; Q1区。
文献一(中文):Tomoko Uehara, Mamiko Yamada, Shuichiro Umetsu等。LSR基因的双等位基因突变导致一种新型的婴儿肝内胆汁淤积症.儿科杂志,2020 Jun;221:251-254.影响因子= 3.739; Q1区。
英文摘要:
We identified biallelic pathogenic mutations in the Lipolysis-stimulated lipoprotein receptor (LSR) gene in a patient with infantile intrahepatic cholestasis. We established that mutations in the LSR gene, which encodes a protein which is critical for the formation of tricellular tight junctions in the liver, are a novel cause of pediatric cholestasis.
中文摘要:
我们在婴儿肝内胆汁淤积患者中发现了脂解刺激脂蛋白受体(LSR)基因中的双等位基因致病突变。LSR基因编码一种对于肝内三细胞紧密连接形成至关重要的蛋白质,LSR基因突变是小儿胆汁淤积的新病因。
丁向春教授简评:
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis, PFIC)是一组以胆汁转运或排泄缺陷为特征的常染色体隐性遗传病,发病率为1/50 000~1/100 000,基因突变导致肝细胞和胆管上皮细胞上与肝内胆汁转运或排泄相关的各种功能蛋白的生成、调控及修饰缺陷是PFIC产生的根本原因,目前根据致病基因不同PFIC分为 6 型,分别由ATP8B1、ABCB11、ABCB4、TJP2、NR1H4 及 MYO5B 基因突变导致。该疾病呈世界性分布,基因突变谱存在种族差异。紧密连接是参与上皮细胞间和内皮细胞间连接的结构。肝细胞之间可通过紧密连接使胆汁中高浓度的物质局限于毛细胆内,不能反流进入血液,肝细胞紧密连接蛋白相关基因突变可导致紧密连接蛋白功能缺陷,当胆汁进入毛细胆管时,由于肝细胞间紧密连接功能异常,胆汁成分可渗漏进入血液,导致肝内胆汁淤积。脂解刺激脂蛋白受体(LSR)可表达在肝脏和其他器官中,是肝内三细胞紧密连接形成至关重要的蛋白质。LSR蛋白功能完全丧失可导致小鼠肝脏发育不全和胎儿死亡,但其在人类健康和疾病中的作用尚未阐明。 Maddirevula S等在2019年曾报道过一例肝内胆汁淤积症患者存在一种罕见的LSR纯合非同义p.Glu235Gly变异,但目前尚不清楚LSR 纯合非同义p.Glu235Gly变异是否可导致肝内胆汁淤积表型,或是否是偶然发生的关联。在另一个具有相同或重叠表型的肝内胆汁淤积患者中显示LSR双等位基因变异可能与患者肝内胆汁淤积相关,但未证实其在患者致病中的因果关系。
本文献报道了一名5岁女性肝内胆汁淤积患者LS R基因(ENST00000361790)的双等位基因突变分析,结果发现该患者与Maddirevula S等在2019年报道的那一例肝内胆汁淤积症患者一样存在LS R基因双等位基因致病变异和肝脏LS R蛋白表达缺乏,证实了LS R缺乏症是婴儿肝内胆汁淤积症的一个新原因,其引起PFIC机制推测与LS R基因致病变异导致肝内三细胞紧密连接蛋白功能缺陷相关。PFIC预后取决于其亚型及基因缺陷的严重程度,本文献报道的病例显示:由LSR基因突变导致的LS R缺乏症所致的肝内胆汁淤积症较其他大多数形式的PFIC发病相对较晚,组织学与纤维化进展相对较轻。LSR基因突变作为小儿胆汁淤积的一种新病因要引起临床专家重视,基因检测是精准诊断PFIC的重要手段。
3.2 文献二(英文):Arend W Overeem, Qinghong Li, Yi-Ling Qiu, et al. A Molecular Mechanism Underlying Genotype-Specific Intrahepatic Cholestasis Resulting From MYO5B Mutations. Hepatology, 2019 Nov 21. Impact factor=14.971; Q1区。
文献二(中文):Arend W Overeem, Qinghong Li, Yi-Ling Qiu等. MYO5B突变导致基因型-特异性肝内胆汁淤积的分子机制.肝脏杂志, 2019 Nov 21. 影响因子=14.971; Q1区。
英文摘要:
BACKGROUND AND AIMS:
Progressive familial intrahepatic cholestasis (PFIC) 6 has been associated with missense but not biallelic nonsense or frameshift mutations in MYO5B, encoding the motor protein myosin Vb (myoVb). This genotype-phenotype correlation and the mechanism through which MYO5B mutations give rise to PFIC are not understood. The aim of this study was to determine whether the loss of myoVb or expression of patient-specific myoVb mutants can be causally related to defects in canalicular protein localization and, if so, through which mechanism.
APPROACH AND RESULTS:
We demonstrate that the cholestasis-associated substitution of the proline at amino acid position 600 in the myoVb protein to a leucine (P660L) caused the intracellular accumulation of bile canalicular proteins in vesicular compartments. Remarkably, the knockout of MYO5B in vitro and in vivo produced no canalicular localization defects. In contrast, the expression of myoVb mutants consisting of only the tail domain phenocopied the effects of the Myo5b-P660L mutation. Using additional myoVb and rab11a mutants, we demonstrate that motor domain-deficient myoVb inhibited the formation of specialized apical recycling endosomes and that its disrupting effect on the localization of canalicular proteins was dependent on its interaction with active rab11a and occurred at the trans-Golgi Network/recycling endosome interface.
CONCLUSIONS:
Our results reveal a mechanism through which MYO5B motor domain mutations can cause the mislocalization of canalicular proteins in hepatocytes which, unexpectedly, does not involve myoVb loss-of-function but, as we propose, a rab11a-mediated gain-of-toxic function. The results explain why biallelic MYO5B mutations that affect the motor domain but not those that eliminate myoVb expression are associated with PFIC6.
中文摘要:
研究的背景和原理:进行性家族性肝内胆汁淤积症6型(PFIC6)与编码运动蛋白myoVb的MYO5B基因的错义突变有关,但与该基因的双等位基因无义突变和移码突变无关。MYO5B突变导致PFIC的基因型-表型相关性和机制尚不清楚。本研究的目的是确定myoVb缺陷或患者特异性myoVb突变体的表达是否与小管蛋白定位缺陷有关,如果有关,是通过哪种机制。
主要结果:我们证明了与胆汁淤积相关的myoVb基因突变 P660L引起了囊泡区胆管蛋白在细胞内积累。值得注意的是,体外和体内敲除MYO5B均不引起小管定位缺陷。相反,仅由尾部结构组成的myoVb突变体的表现了Myo5b-P660L突变的作用。使用myoVb和rab11a的突变体,我们证明了运动域缺陷型myoVb抑制了特殊的尖端回收内体的形成,其对小管蛋白定位的破坏取决于其与活性rab11a的相互作用,该作用发生在反式高尔基体网络/回收内体界面。
结论:我们的结果揭示了一种机制,MYO5B运动域突变导致rab11a介导的毒性增加,从而引起肝细胞中小管蛋白的错误定位,但不导致myoVb功能丧失。这就解释了影响运动域MYO5B双等位基因突变导致PFIC6,但又不完全抑制myoVb表达。
丁向春教授简评:
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis, PFIC)根据致病基因不同PFIC分为 6 型,分别由ATP8B1、ABCB11、ABCB4、TJP2、NR1H4 及 MYO5B 基因突变导致。其中PFIC6与编码运动蛋白myoVb的MYO5B基因的错义突变有关,但与该基因的双等位基因无义突变和移码突变无关。MYO5B突变导致PFIC的基因型-表型相关性和机制尚不清楚。
为进一步明确MYO5B基因突变在PFIC6发病作用和机制,本研究作者首先证明了与胆汁淤积相关的myoVb基因突变 P660L是引起了囊泡区胆管蛋白在细胞内积累的原因,但作者发现体外和体内实验发现敲除MYO5B并不会引起小管定位缺陷,仅检测到由尾部结构组成的myoVb突变体的表现了Myo5b-P660L突变的作用。最后本研究作者使用myoVb和rab11a的突变体实验证明运动域缺陷型myoVb抑制了特殊的尖端回收内体的形成,其对小管蛋白定位的破坏取决于其与活性rab11a的相互作用,该作用发生在反式高尔基体网络/回收内体界面。本研究中作者揭示PFIC6发病的一种新的机制,即:MYO5B运动域突变可导致rab11a介导的毒性增加,从而引起肝细胞中小管蛋白的错误定位,但不导致myoVb功能丧失。胆盐外运泵(bile salt export pump, BSEP) 是肝细胞毛细胆管膜上的胆盐转运蛋白及肝细胞毛细胆管面分泌胆汁酸的运载体,目前在人类尚未发现替代途径。人类胆汁流的形成 75% 是胆盐依赖性的,胆盐是形成胆汁流的主要驱动力。BSEP表达水平下降或功能损害均会影响胆盐分泌,影响胆汁流形成,导致胆汁淤积, 蓄积的胆汁酸对肝细胞造成毒害,引起炎症和纤维化。BSEP只在肝细胞中表达,其参与的过程是胆汁的肠肝循环中的关键步骤,其功能主要是将胆汁逆浓度梯度转运出肝细胞。MYO5B与Ras相关蛋白Rab-11a的相互作用对编码BSEP定位到小管膜至关重要。本研究作者发现MYO5B 缺陷导致rab11a介导的毒性增加并可引起肝细胞中小管蛋白的错误定位,为临床应用熊去氧胆酸、部分胆汁外分流术和部分胆汁内分流术治疗PFIC6提供理论基础。
4.本期Pubmed文献编译及短评专家简介
4.1 文献编译

白洁:首都医科大学附属北京佑安医院在读博士研究生
佑安专科联盟遗传代谢性肝病专业委员会秘书
邮箱:docbai@yeah.net
4.2 短评专家
丁向春:男,主任医师,教授,博士研究生导师
宁夏医科大学总医院
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
三、 临床资讯
1.典型病例分享
1.1 病例简介(PFIC-3):
患者男性,15岁,主因“尿黄1年,发现肝硬化2周”于2019-08-22收入我科。
患者1年前无明显诱因出现尿黄,偶有厌油、皮肤轻度瘙痒,无明显腹胀、腹痛、食欲减退及皮肤、巩膜黄染等情况,未予重视 。2周余前(2019-08-06),家属发现患者出现皮肤、巩膜黄染,遂就诊于当地医院。行肝功能检查:ALT 148U/L,AST 18U/L, ALB 32.3g/L,TBIL 101μmol/L,ALP 333U/L,GGT 397U/L;血常规检查:WBC 4.12×109/L,HGB 124g/L,PLT 65×109/L。腹部CT检查示:肝硬化、脾大、侧支循环形成。胃镜提示:食管胃底静脉曲张 重度,门脉高压性胃黏膜病变。骨穿检查结果:骨髓增生大致正常,红系比增高;余未见异常。肝组织活检病理诊断:中度慢性肝炎G3S4。眼科行眼底检查未见K-F环,当地医院未予明确诊断,经保肝、降酶等治疗后,效果欠佳。现患者为行进一步诊治,收入我科。
患者自发病以来精神可,食量无变化,睡眠无改变,尿液呈深黄色,尿量无改变,大便正常,体重无变化。
既往史:对青霉素过敏。余无特殊。
家族史:父母体健,有2个姐姐,母亲和二姐有胆囊结石病史。大姐曾诊断妊娠期肝内胆汁淤积。否认肝病家族史、肿瘤家族史及其他遗传性疾病家族史。
入院查体:T 36.5℃,P 82次/分,R 22次/分,BP 105/62mmHg。慢性面容,皮肤、巩膜中度黄染,未见肝掌、蜘蛛痣及毛细血管扩张,未触及明显浅表淋巴结肿大。心肺无明显异常。腹软,无压痛、反跳痛及肌紧张,肝肋下未及,脾大,脾脏右侧边缘可达正中线,下缘位于脐下2cm。移动性浊音阴性,肠鸣音4次/分,Murphy征阴性,双下肢无水肿。神经系统查体未见异常。
入院初步诊断:肝硬化原因待查 遗传代谢性疾病?脾功能亢进 食管胃底静脉曲张 门脉高压性胃病 肝门区、腹膜后多发淋巴结增大
入院检查结果(2019-08-23,佑安医院):
血常规:WBC 3.41×109/L, HGB 121g/L, PLT 77×109/L。
肝功能+血生化+血脂:ALT 139.5U/L, AST 183.7U/L, TBIL 107.7μmol/L, DBIL 77.5μmol/L, ALB 32.9g/L, ALP 348U/L, GGT 394.3U/L, TBA 115.5μmol/L,CHE 2879U/L,Crea 28.5μmol/L, eGFR 193.53ml/min/1.73m2, K 3.99mmol/L, Na 140.3mmol/L, Cl 107.4mmol/L,TG 1.12mmol/L,CHOL 4.46mmol/L。
凝血象:PT 13.6s, PT% 75%, INR 1.21, APTT 37.8s。
特种蛋白:IgG 19.7g/L, IgA 4.46g/L, IgM 3.46g/L, 转铁蛋白 1.96g/L,α1-抗胰蛋白酶 2.05g/L,抗链球菌溶血素“O”447IU/mL,铜蓝蛋白正常。
IgG亚类:免疫球蛋白G1 17.7g/L,其余正常。
自身抗体系列:ANA 1:100 核颗粒,ASMA 1:100。
T细胞亚群:未见异常;甲状腺功能正常。
性激素六项:雌二醇、卵泡刺激素、促黄体生成素、泌乳素及睾酮正常,孕酮:0.7nmol/L。
病毒学检查:甲型、乙型、丙型、戊型、CMV、EBV检查均为阴性。
肝脏弹性测定:CAP(dB/m)中值:236;E(kPa)中值:49.6。
心电图及心脏彩超:大致正常。胸部平扫CT:未见明确异常。
MRCP检查:胆囊炎,胆管炎可能。
上腹部增强CT(图1):肝脏炎性改变可能;肝硬化,脾大,侧支循环形成;脾内多发低密度灶,建议MR检查;腹腔多发肿大淋巴结,炎性增生反应可能,建议复查。
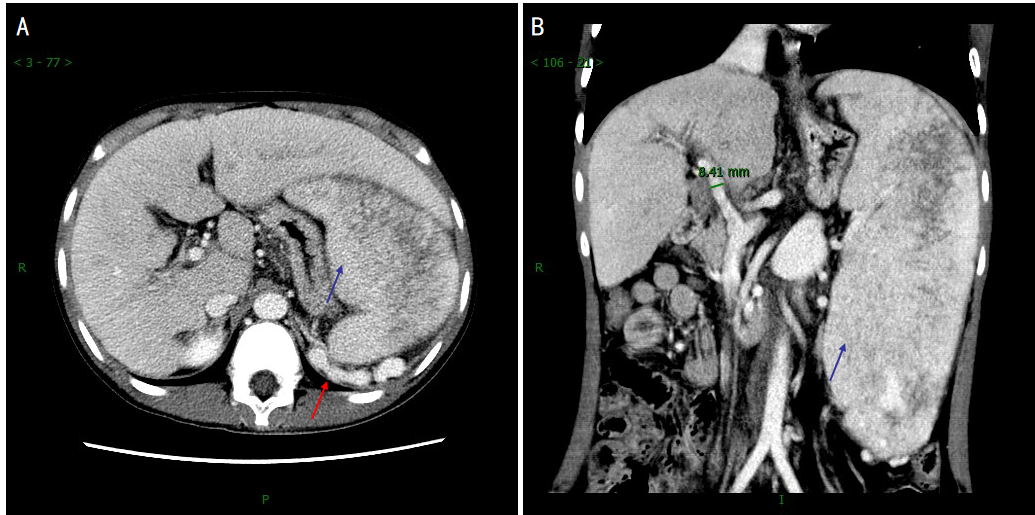
图1.上腹部CT
胃镜(图2)提示食管静脉曲张重度:
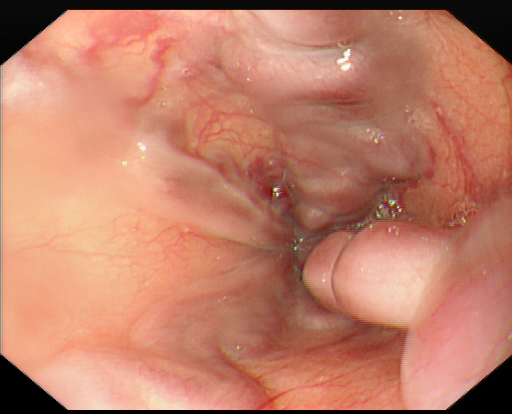
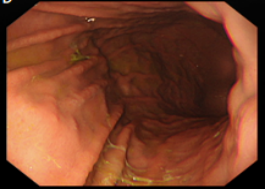
图2.电子胃镜
我院病理会诊意见:
(肝穿)慢性肝炎,G2-3S4,伴有淤胆(图3)。免疫组化:ck19(部分胆管消失),ck7(慢性胆盐淤积),mum-1(少量+)。特染:Tim(+)。
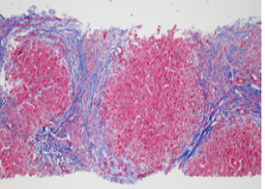
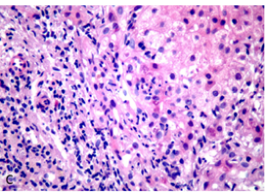
图3.病理染色 Masson 100x/H&E 400x
患者巨脾,轻度贫血,血小板降低,请外院血液科会诊,再次行骨穿+活检检查。骨穿结果回报:红系增生明显活跃。血液科意见首先考虑:肝硬化,脾亢。建议积极治疗肝病,监测淋巴结情况,必要时活检。
患者住院期间出现发热,Tmax 37.5℃,考虑胆囊炎,予头孢唑肟抗感染治疗。予复方甘草酸苷、多烯磷脂酰胆碱、谷胱甘肽保肝,丁二磺酸腺苷蛋氨酸退黄,熊去氧胆酸促进胆汁淤积排泄,前列地尔和丹参酮改善肝脏微循环等治疗2周后,复查肝功能示(2019-09-06,我院):ALT 83U/L, AST 127U/L, TBIL 182.7μmol/L, DBIL 135.2μmol/L, ALB 29.7g/L, ALP 413U/L, GGT 244U/L, TBA 138.1μmol/L,CHE 2408U/L。患者转氨酶较前下降,胆汁淤积未见明显改善。抗感染基础上,间断给予激素(甲强龙静点)后,总胆红素可短暂下降,后又持续升高。
基因检测结果:患者ABCB4基因c.T2525C(p.L842P)和c.T3152C(p.V1051A)双位点杂合突变,不除外PFIC-3型。患者两个姐姐和患者存在相同突变位点。患者和二位姐姐的两个突变位点分别来自母亲(c.T2525C)和父亲(c.T3152C)。进一步绘制家系图(图4)
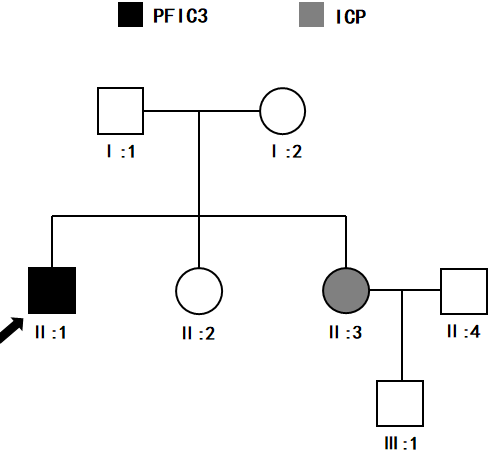
图4. 家系图
结合家族史和患者病史,考虑PFIC-3型可能性大,预后差。继续给予保肝利胆治疗,患者于2019-10-15病情好转出院。
诊治体会:患者15岁,1年前开始出现黄疸,生化提示转氨酶轻度升高,胆红素明显升高,以直接胆红素升高为主,TBA、ALP和GGT均明显升高,影像学提示肝硬化,未见胆道梗阻表现。病理提示肝细胞损伤较轻,纤维化重,G2-3S4,伴有淤胆。故临床诊断胆汁淤积性肝硬化明确。患者青少年男性,主要表现为GGT升高的肝内胆汁淤积。基因检测发现ABCB4双位点杂合突变,进一步完善家系基因检测发现 :其二位姐姐和患者携带相同的突变位点,三个孩子的突变位点分别来自父亲和母亲(即复合杂合突变)。ABCB4基因突变可引起良性复发性肝内胆汁淤积症(Benign recurrent intrahepatic cholestasis,BRIC)、妊娠期肝内胆汁淤积症(Intrahepaticcholestasis of pregnancy,ICP)、药物诱发的胆汁淤积(Drug induced cholestasis,DIC)和低磷脂相关性胆石症(Low-phospholipid-associated cholelithiasis,LPAC)等。这些疾病表现形式不同,轻度不等的表型,实际上代表相同疾病的连续体。性别、激素水平、多种胆汁淤积相关基因的变异、表观遗传调控和环境等均可能是影响表型(临床表现)差异的原因。本患者家族史中,其大姐曾出现妊娠期肝内胆汁淤积表现,生产后好转;其母亲和二姐有胆囊结石病史。两位姐姐和母亲均有发病,但表现形式不同,病情相对较轻。患者15岁出现肝硬化,病情进展很快,治疗过程中对常规保肝退黄等治疗反应欠佳,PFIC-3型诊断成立,预后差。
本病例提示我们:PFIC因常发病年龄较早而多在儿科诊断。临床上对于不明原因的肝硬化儿童或青少年患者,当合并胆汁淤积症时,应考虑到家族性胆汁淤积可能,及时启动基因检测往往有助于诊断。建议送检标本做好包括一代亲属,尤其父母亲同时送检,有助于通过家系共分离分析而增加诊断效能。
1.2 专家点评
丁向春教授:进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis, PFIC)是一组以胆汁转运或排泄缺陷为特征的常染色体隐性上遗传病,发病率为1/50 000~1/100 000,基因突变导致肝细胞和胆管上皮细胞与肝内胆汁转运或排泄相关的各种功能蛋白的生成、调控及修饰缺陷是PFIC产生的根本原因,目前根据致病基因不同PFIC分为6型。PFIC-3型是ABCB4基因突变所致。ABCB4基因定位于染色体 7q21区域,编码多重耐药蛋白 3(multidrug resistance protein 3, MDR3)。MDR3是位于肝细胞毛细胆管膜的磷脂输出泵,将磷脂从肝细胞转运到胆管,是磷脂分泌的限速步骤。正常情况下,肝细胞合成的磷脂通过MDR3转运到胆汁中,与胆盐共同形成微粒,使胆盐亲水性增加,减轻胆盐的去垢作用,保护胆管细胞免受胆盐的毒性损伤。ABCB4基因突变可导致MDR3表达减少或功能损害,胆汁中磷脂减少或缺乏,游离胆盐直接对毛细胆管膜发生毒性去垢作用,损伤胆管细胞,出现胆汁淤积等。根据 ABCB4基因突变致MDR3功能损害严重程度不同,分为PFIC3型或BRIC3。在成年人,可表现为低磷脂性胆石症或妊娠时肝内胆汁淤积症。
PFIC诊断需要在综合家族史、临床症状及体征、实验室生化检测、胆汁分析、影像学及病理学检查的基础上加以基因分子生物学分析来判断。
本例患者男性,15岁,尿黄1年,皮肤轻度瘙痒,因发现皮肤、巩膜黄染2周余,当地肝功能检查表现为黄疸以直接胆红素增高为主,ALP和GGT高;血常规检查PLT减低;胃镜显示食管胃底静脉曲张 重度,门脉高压性胃黏膜病变;骨穿结果显示骨髓增生大致正常,红系比例增高;肝组织活检病理显示G3S4;未见K-F环,当地医院肝硬化原因未能明确。本次住院期间追问病史发现患者母亲和二姐有胆囊结石病史,大姐曾诊断过妊娠期肝内胆汁淤积。由此特点考虑患者肝硬化原因可能为遗传代谢性疾病,并进行了病毒性肝炎、自身免疫性肝病、铜蓝蛋白、血清铁、铁蛋白、总铁结合力等排查,结果均阴性。随后对当地医院的肝组织病理进行了会诊显示为慢性肝炎,G2-3S4,伴有淤胆;免疫组化:ck19(部分胆管消失),ck7(慢性胆盐淤积),mum-1(少量+);特染:Tim(+)。再次骨穿结果考虑:肝硬化,脾亢。为了明确病因进行基因检测显示患者ABCB4基因c.T2525C(p.L842P)和c.T3152C(p.V1051A)双位点杂合突变,不除外PFIC-3型。患者两个姐姐和患者存在相同突变位点,且患者和二位姐姐的两个突变位点分别来自母亲(c.T2525C)和父亲(c.T3152C)。该病例结合患者病史、家系史及临床特点,最终经基因检测诊断PFIC-3型成立。
有益提示:PFIC确诊依赖于基因测序技术,今后临床工作中应重视不明原因肝硬化尤其是存在肝内胆汁淤积的患者的基因检测。对于PFIC患者要重视家系发病追溯,另外还应注意排除胆道闭锁、Alagille综合征、a-抗胰蛋白酶缺乏症、囊性纤维化、硬化性胆管炎和肝外梗阻等其疾病的可能性。PFIC发病机制如需进一步研究明确。
1.3 病例供稿及点评专家简介
1.3.1供稿专家:

李璐:博士,主治医师
首都医科大学附属北京佑安医院疑难肝病及人工肝中心
白洁:首都医科大学附属北京佑安医院在读博士研究生
佑安专科联盟遗传代谢性肝病专业委员会秘书
邮箱:docbai@yeah.net
1.3.2 点评专家:丁向春教授
丁向春:男,主任医师,教授,博士研究生导师
宁夏医科大学总医院
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
四、 专业委员会名单(按拼音排序):
白洁、白丽、包双宝、边巴央珍、卞丹丹、曾惜秋、常乐、陈芳、陈连清、陈琳、陈悦、程孟怀、程全红、次仁、代东旺、邓雪梅、丁建强、丁向春、杜方雄、段宏宪、段雪飞、段钟平、冯铁柱、高珍、葛迎春、巩维进、苟卫、顾伟玲、郭宁、郭文征、郭小青、何云、胡善雷、黄明星、黄祖雄、霍丽亚、江守伟、焦洪波、经继生、鞠莹、孔明、李宝生、李博、李灿、李广明、李红玲、李建国、李娟、李军、李俊峰、李磊、李秋莲、李荣宽、李响、刘彩峰、刘菲菲、刘晖、刘梅、刘霜、刘冉、刘晓彦、刘耀敏、刘振中、罗磊、吕帅、马玉秀、马臻、苗艳艳、牟丹蕾、南月敏、宁寒冰、牛卫理、彭鹏、乔晓红、邵鸣、盛云建、宋正已、苏丽、孙美艳、覃丽华、谭林、王德步、王飞、王健、王建设、王丽华、王小凤、王艳巧、王仲培、吴刚、吴万锋、吴晓枫、向光明、肖玉珍、谢秋里、易永芬、杨艳玲、于德顺、袁喜先、张定琳、张帆、张立婷、张丽、张丽娟、张缭云、张妍、张银华、张玉山、张月荣、张志刚、张宗超、赵守松、赵素贤、郑素军、周晓丽、周晓玲、邹桂舟
五、 联系方式
投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
联系电话:010-63291007
联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间