
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会
中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张敏
执行编辑:郑素军,汤珊,侯维
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝
病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
肝豆状核变性(hepatolenticular degeneration,HLD)又名威尔逊病(Wilson Disease, WD),是位于人类13号染色体q14.3的ATP7B基因突变造成编码铜转运P型ATP酶功能缺陷或丧失,导致肝细胞内铜输出缺陷,胆道排铜障碍,铜大量蓄积于肝、脑、肾、骨关节、角膜等组织和脏器,患者出现肝脏损害、神经精神表现、肾脏损害、骨关节病及角膜色素环(Kayser-Fleischer ring,K-F 环)等表现。WD欧美报道发病率大约1:30000,我国约1:10000, 由于携带频率约为 1/90,推算发病率应在1:7194。Arg778Leu是中国人Wilson病的突变热点。
杜兴型肌营养不良(Duchenne muscular dystrophy,DMD)是由于位于X染色体上的抗肌萎缩蛋白(Dystrophin,dys)基因突变所致的一种X染色体连锁隐性遗传疾病,多发生于男孩,发生率大约1:3500个新生男婴,女性则需两条X染色体均出现突变才会发病。DMD患者的发病机制是由于dys缺乏导致了骨骼肌细胞膜缺陷,细胞内的肌酸激酶等外漏,肌细胞坏死、脂肪组织和纤维结缔组织增生。DMD早期的主要表现为下肢近端和骨盆带肌萎缩和无力、小腿腓肠肌假性肥大、鸭步和Gowers征,晚期可出现全身骨骼肌萎缩,通常在20多岁死于呼吸衰竭或心力衰竭。DMD 65%的病例是遗传而来,35%是基因突变而来。
一个患者同时发生这两种罕见疾病的概率极低。本期月报报告一例肝豆状核变性合并杜氏肌营养不良的7岁患儿,主因发现肝功异常3年就诊,从临床上既有肝豆状核变性导致的肝损害,又伴有明显的肌损害征象。为了解其精准诊断及基因变异来源,我们对患儿进行了基因检查,同时对其做了尽可能详细的家系调查。

张敏
医学博士,主任医师,副教授
解放军总医院第五医学中心肝病科三病区主任
中华医学会肝病学分会遗传代谢性疾病学组委员
中华医学会医学遗传学分会生化和代谢学组委员
北京医学会遗传代谢病分会委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
■马拉利昔布治疗 Alagille 综合征和胆汁淤积性瘙痒症 (ICONIC) 患者的疗效和安全性:一项随机 2 期研究(Lancet,2021, IF=60.392; Q1区)

■NOTCH2突变引起的Alagille综合征呈现非典型病变(Clin Chim Acta,2021, IF=2.615; Q2区)
■遗传性代谢性肝病患儿的神经系统发育(J Pediatr Gastroenterol Nutr ,2021, IF=2.937; Q1区)
■金属硫蛋白免疫组化对Wilson病的检测具有较高的敏感性和特异性(Mod Pathol ,2021, IF=5.988; Q1区)
■威尔逊病小鼠模型中出现肝脏脂肪变性(Am J Pathol ,2022, IF=3.491; Q1区)
■外泌体MicroRNA可作为Ia型糖原贮积病患者肝损伤和肾脏疾病的潜在生物标志物(Int J Mol Sci ,2021, IF=4.556; Q2区)
■非靶向脂质组学揭示了威尔逊病的特征性脂质特征(Front Pharmacol,2021, IF=5.81; Q2区)
■综述:庞贝病的药物疗法(Molecules ,2021, IF=3.267; Q2区)
■威尔逊病患者的营养状况分析:来自中国的横断面研究(Front Nutr,2021, IF=3.365; Q2区)
■双胆碱四硫钼酸盐在Atp7b缺陷铜过载小鼠模型中的治疗效果(Biomedicines,2021, IF=4.717; Q2区)
三、临床资讯
1.病例分享
患儿男性,7岁,因“发现转氨酶高、步态异常3年余”就诊。4岁时体检发现肝功异常,ALT200U/L,间断复查仍异常,ALT波动于70-200U/L,同时有运动能力差,行走时步态异常,走路易摔倒,并逐渐加重。为诊治肝功异常来我院。
个人史:患儿系第2胎,足月顺产,出生体重2.7Kg。母乳喂养至1岁半,运动能力发育较差,走路易摔倒,且每次摔倒头先着地。无血吸虫病疫水接触史,无放射性物质、毒物接触史,无其他特殊嗜好。
既往史及家族史:患者2岁后因“扁桃体炎”常应用头孢类抗生素、蒲地兰口服液等治疗,每年发作4-5次,每次用药约1周。否认肝炎等传染病史,否认“高血压”等病史,否认外伤、手术史,否认输血史,否认药物、食物过敏史,按国家计划预防接种。父母均体健,父母为近亲结婚(患儿的奶奶与其姥爷是亲兄妹)。患儿表兄(母亲的孪生姐姐之子)亦转氨酶高,且肌酸激酶>10000U/L。
入院查体:全身皮肤、巩膜无黄染,未见瘀斑、出血点。肝掌阳性,蜘蛛痣阴性。头颅五官无畸形,甲状腺无肿大,心肺阴性。全腹无压痛、反跳痛,肝脾肋下未触及。走路步态不稳,翼状肩胛,双侧腓肠肌肥大,肌力检查:颈曲2级,上肢内收3级,下肢内收、伸膝2级,屈髋、背伸3级,跟腱挛缩,腱反射减低。Gowers征阳性。
入院后化验检查
生化指标:ALT 386U/L、AST 231U/L、CK 9288U/L、CK-MB 207.4ng/ml、铜蓝蛋白CER 0.02g/L、24小时尿铜:105.8ug(参考值15-30)。
血常规:血小板 287×10^9/L、中性粒细胞百分比 68.14%、白细胞 8.91×10^9/L、血红蛋白 114g/L。
甲胎蛋白2.22ng/ml。
血氨 24.2umol/L、乳酸 1.38mmol/L。
甲状腺功能正常。
甲乙丙戊肝炎病毒标志物、巨细胞病毒IgM抗体、EB 病毒IgM抗体、EB病毒早期抗原IgM抗体均阴性。CMVDNA <100IU/ml、血浆EBVDNA<100IU/ml。
自身抗体14项、抗核抗体谱:均阴性。
辅助检查
腹部超声:肝回声增粗、脾大。
眼科检查:双眼角膜K-F环阴性。
头颅MRI:未见异常。
心电图:窦性心律 正常范围心电图。
胸片:双肺未见明确病变。
心脏超声:三尖瓣少量反流
肝脏硬度值:4.5Kpa。
肝穿病理:考虑遗传代谢障碍性肝病,肝豆状核变性可能性大G2S2-3;铜染色(+)。
双大腿MRI:双侧臀大肌及大腿肌肉脂肪浸润伴多发水肿改变。
左肱二头肌病理诊断:骨骼肌呈肌营养不良样病理改变。
全外显子基因检测
结果:在肝豆状核变性相关基因ATP7B存在两处纯合突变(ATP7B c.2333G>T chr13:52532469 p.R778L纯合突变-已知致病突变;ATP7B c.2310C>G chr13:52532492 p.L770L纯合突变-疑似致病突变),家系验证结果显示此两处纯合突变分别来自于其父母(图1)。同时发现该样本DMD基因51号外显子有缺失突变。
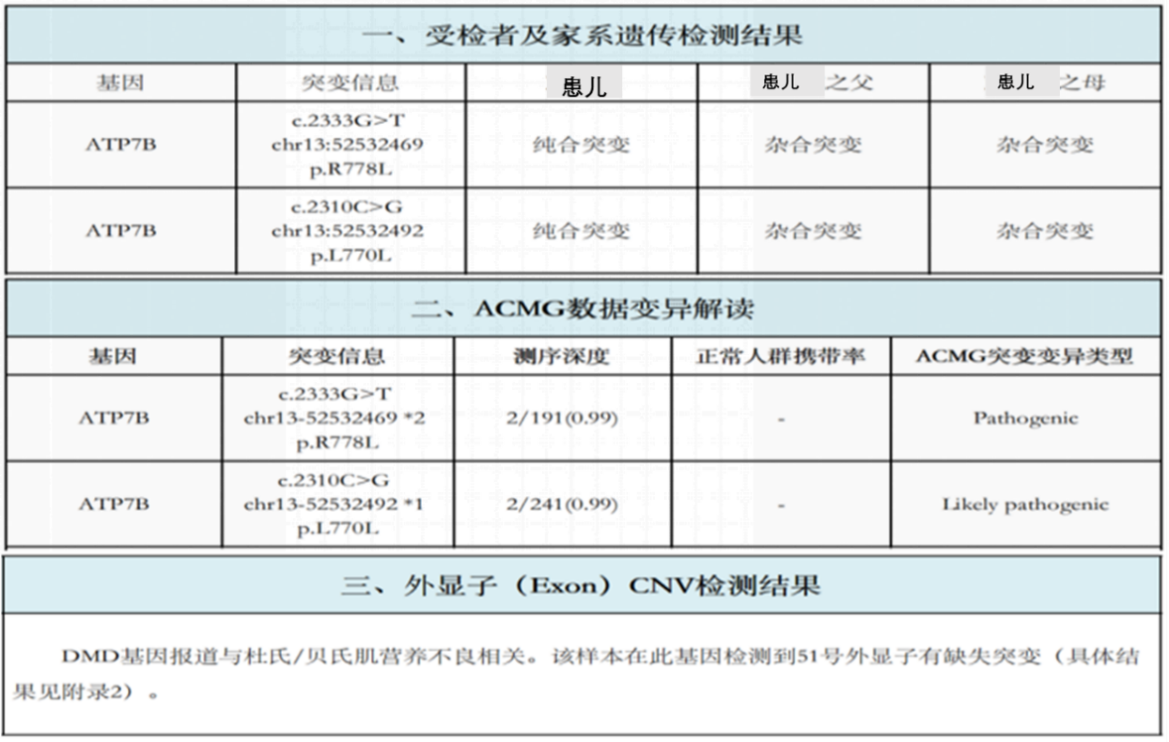
图1 患儿及父母全外显子检测报告
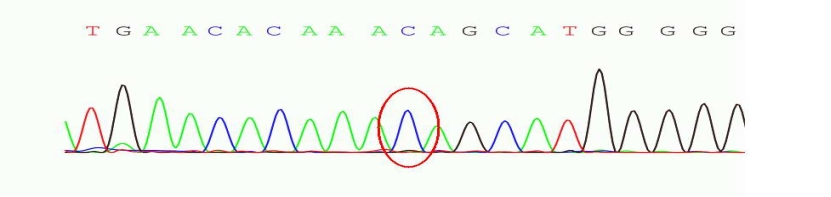
图2 chr13:52532492存在 c.2310C>G 的纯合突变
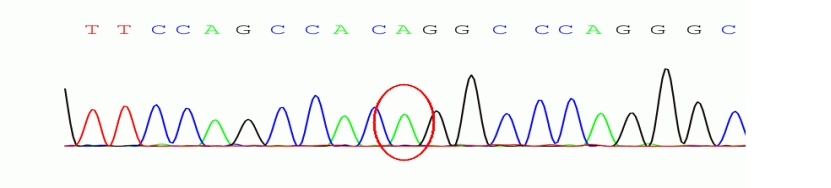
图3 chr13:52532469存在 c.2333G>T 的纯合突变
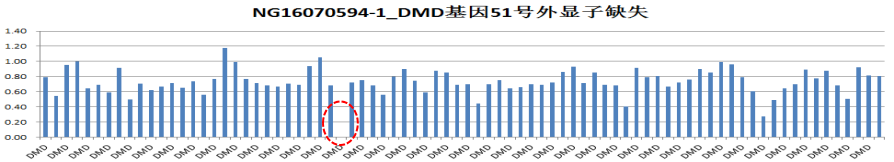
图4 DMD基因51号外显子缺失
患儿基因检测结果ATP7B基因两个纯合突变:c.2310C>G;c.2333G>T,分别来自父亲和母亲。推断两个变异位点可能为该患儿的致病性突变,临床表型为肝豆状核变性;基因检测同时存在DMD基因51号外显子缺失,临床表型为DMD。根据患儿临床表现、查体、化验指标、辅助检查及基因检测,符合肝豆状核变性、杜兴型肌营养不良症诊断。为证实诊断进一步做父母及家系验证。
父母及家系验证
行父亲、母亲、姐姐基因验证。因通过详细问诊做家系上溯4代调查,发现有近亲结婚及多例肝豆状核变性或/和DMD患者,尽可能动员表兄、小姨、舅舅行基因验证,其ATP7B c.2333、ATP7B c.2310、DMD基因51号外显子3个结果如图5-7所示。
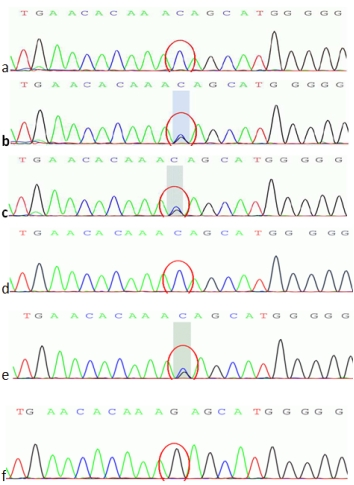
图5 ATP7B c. 2310C>G 二代测序图
a. 先证者; b.先证者母亲;c.先证者父亲;
b. d.先证者姐姐; e.先证者小姨; f.先证者舅舅
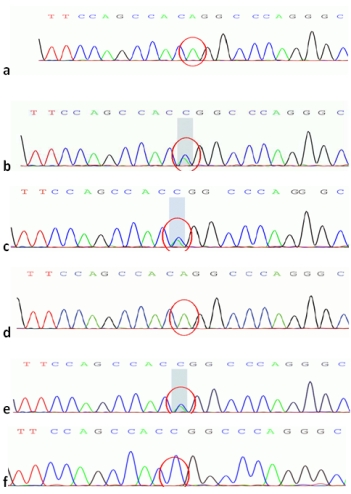
图6 ATP7B c. 2333G>T 二代测序图
a. 先证者; b.先证者母亲;c.先证者父亲;
b. d.先证者姐姐; e.先证者小姨; f.先证者舅舅
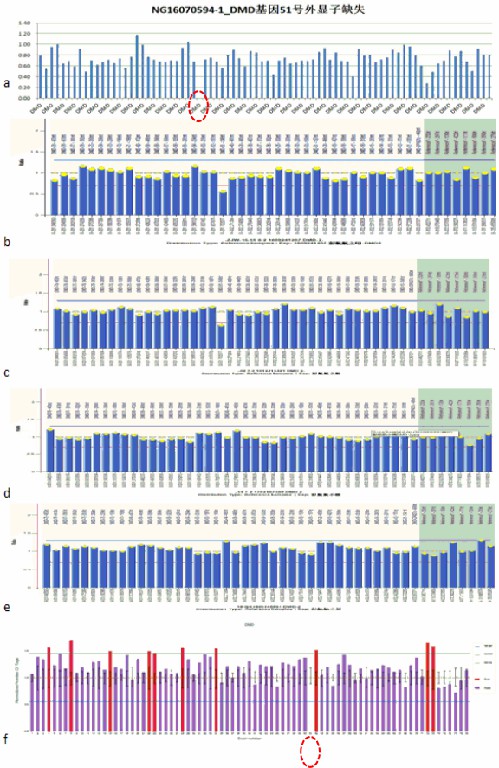
图7 DMD基因51号外显子测序图
a. 先证者; b.先证者母亲; c.先证者姐姐;
b. d.先证者小姨; e.先证者舅舅; f.先证者表哥
基因验证结果显示先证者母亲(Ⅲ-3)存在ATP7B基因单杂合双突变及DMD基因51号外显子的杂合缺失变异;先证者父亲(Ⅲ-2)存在ATP7B基因单杂合双突变,无DMD基因51号外显子的缺失重复变异,临床表型均正常;先证者的姐姐Ⅳ-1存在与其相同位点的ATP7B基因纯合突变,临床诊断为肝豆状核变性,同时存在DMD基因51号外显子的杂合缺失变异,为DMD女性携带者。
先证者大姨(Ⅲ-4)是其母亲(Ⅲ-3)的同卵双生姐姐(外观推测),未行基因验证,临床表型正常。其儿子(先证者表哥Ⅳ-3)亦转氨酶高,且伴肌酸激酶显著升高,有DMD基因51号外显子缺失突变,未发现ATP7B基因突变,外院已确诊为杜氏肌营养不良症。
先证者舅舅Ⅲ-7未发现ATP7B基因突变及DMD基因51号外显子的缺失重复变异,表型正常;其小姨Ⅲ-4存在ATP7B基因单杂合双突变,未见DMD基因51号外显子的缺失重复变异,临床表型正常。
家系调查
通过基因验证又发现家族内多例肝豆状核变性或/和DMD患者及携带者,所以通过详细问诊做家系上溯4代调查。先证者父母为近亲结婚,先证者的奶奶Ⅱ-2与其姥爷Ⅱ-8是亲兄妹。先证者母系Ⅱ代亲属中有三人50岁左右死于不明原因肝硬化,具体原因不详。根据问诊及基因验证画出家系图谱 (如图8)
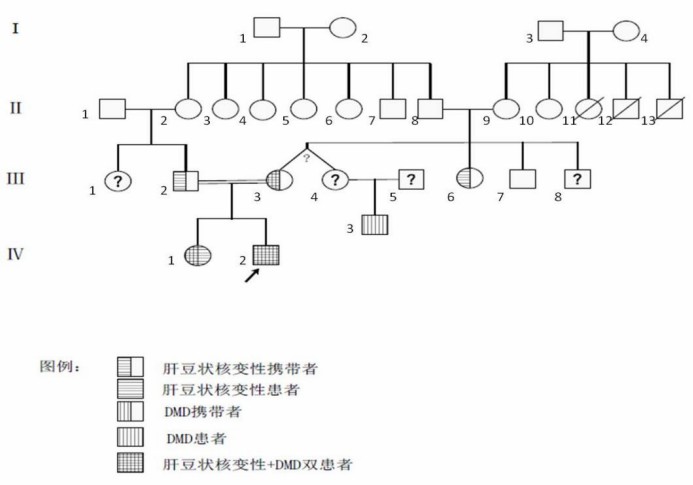
图8. 家系图
家系图注释:
IV-1为先证者姐姐,WD患者及DMD携带,IV-2为先证者,WD+DMD确诊者
Ⅲ-2为先证者父亲,携带WD基因,很可能来源于父系家族Ⅱ-2(先证者奶奶),Ⅲ-3为先证者母亲,携带WD基因和DMD基因,Ⅲ-4为先证者大姨,与Ⅲ-3同卵双生,理论上基因相同,亦为携带WD基因和DMD基因两种致病突变。Ⅲ-6(小姨)仅携带ATP7B突变,未携带DMD,Ⅲ-7(舅舅)未携带任何致病基因,因此推测母系Ⅲ代的ATP7B突变来自Ⅱ-8姥爷,Ⅲ-3、Ⅲ-4的DMD基因异常均源自Ⅱ-9姥姥
Ⅱ-8为先证者姥爷,携带ATP7B突变,Ⅱ-9为先证者姥姥,携带DMD基因异常,是否携带ATP7B突变不能确定。通过先证者母系Ⅱ代亲属中有三人50岁左右死于不明原因肝硬化,推测Ⅱ-9家族除有DMD致病基因,可能还有ATP7B突变,但未行检测,不能确认。如果确实如此,I-4女性为两病携带者或DMD携带+肝豆状核变性患者,I-3男性则可能携带ATP7B突变基因。
同一家族内近亲通婚使先证者ATP7B的致病突变呈现单基因双突变的纯合。庆幸的是肝豆状核变性的表型并未表现非常严重,得以早发现,早治疗。但母系家族携带的DMD致病基因导致男性患儿出现了DMD临床表型。
治疗及随访
在肝豆状核变性治疗方面,给予青霉胺片、二巯丁二酸胶囊排铜,硫酸锌片抑制铜吸收;予复方甘草酸苷、还原型谷胱甘肽保肝降酶等治疗。经排铜等治疗,ALB 35-40g/L左右、CHE 4500-5000U/L、PTA基本正常,肝纤维化未见进一步进展,未出现语言障碍等神经系统症状。
在肌营养不良治疗方面,给予高蛋白饮食,补充乳清蛋白、左旋肉碱、精氨酸,口服甲泼尼龙片、果味钾、沙丁胺醇、二甲双胍、钙剂等;嘱其减少活动,每日泡脚、按摩,康复训练。随访至今患者ALT仍波动于300U/L左右,考虑与肌损伤相关;CK 及CK-MB无下降,肌力逐渐下降,随访至患儿12岁,已无法行走。
分析讨论要点
1.本例先证者通过基因检测证实肝豆状核变性诊断,为ATP7B c.2333G>T chr13:52532469 p.R778L和c.2310C>G chr13:52532492 p.L770L的双位点纯合突变。前者为已知致病突变,后者为疑似致病突变。近亲结婚可能是患儿出现纯合突变的原因。
2.先证者母系Ⅱ代亲属中Ⅱ-11、Ⅱ-12、Ⅱ-13因肝硬化50岁死亡,应考虑肝豆状核变性可能,有条件可在Ⅱ代做基因验证。
3.先证者DMD基因51号外显子存在缺失突变;其母亲、姐姐均存在相同位点的杂合缺失变异,为DMD携带者;表哥也确诊DMD。表明IV代的DMD为遗传而来,并非突变。
4.经家系分析推算I代父系家庭中含有ATP8B1致病突变,I代母系家族中含有女性DMD基因51号外显子缺失突变,不除外携带ATP8B1致病突变。
5.先证者母亲及其母亲的孪生姐姐Ⅲ-4再次生育男婴患DMD概率高达50%。为防止IV代女性子女的发病,强烈建议遗传咨询及产前检查。
6.由于杜兴型肌营养不良尚无有效治疗方法,故高效、准确的基因检测有助于产前诊断及遗传咨询,为预防本病提供关键的技术支持。随着基因编辑技术的快速进展,在基因诊断的基础上进行有效的基因治疗成为可能,也许为先天遗传性疾病的治疗带来希望。
2 幻灯分享
2.1标题及链接:一个肝豆状核变性合并杜兴型肌营养不良症的家系报道
1.2供稿专家简介
张敏
医学博士,主任医师,副教授
解放军总医院第五医学中心肝病科三病区主任
中华医学会肝病学分会遗传代谢性疾病学组委员
中华医学会医学遗传学分会生化和代谢学组委员
北京医学会遗传代谢病分会委员
四、联系方式
■投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
■联系电话:010-63291007
■联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间