
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:赵鸿
执行编辑:郑素军,汤珊,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
卟啉病(porphyria)是血红素合成途径中酶活性缺陷引起的代谢障碍性疾病,由卟啉和/或其前体物质在皮肤、肝脏、神经系统等部位沉积所致。根据发生基因突变的酶种类可分为8种类型(见表1),突变类型包括纯合型、杂合型等,可为遗传性或获得性。根据主要临床表现可将卟啉病分为皮肤型(PCT、CEP、EPP、XLPP)、神经症状型(AIP、ADP)及混合型(VP、HCP)。
表1. 卟啉病分型
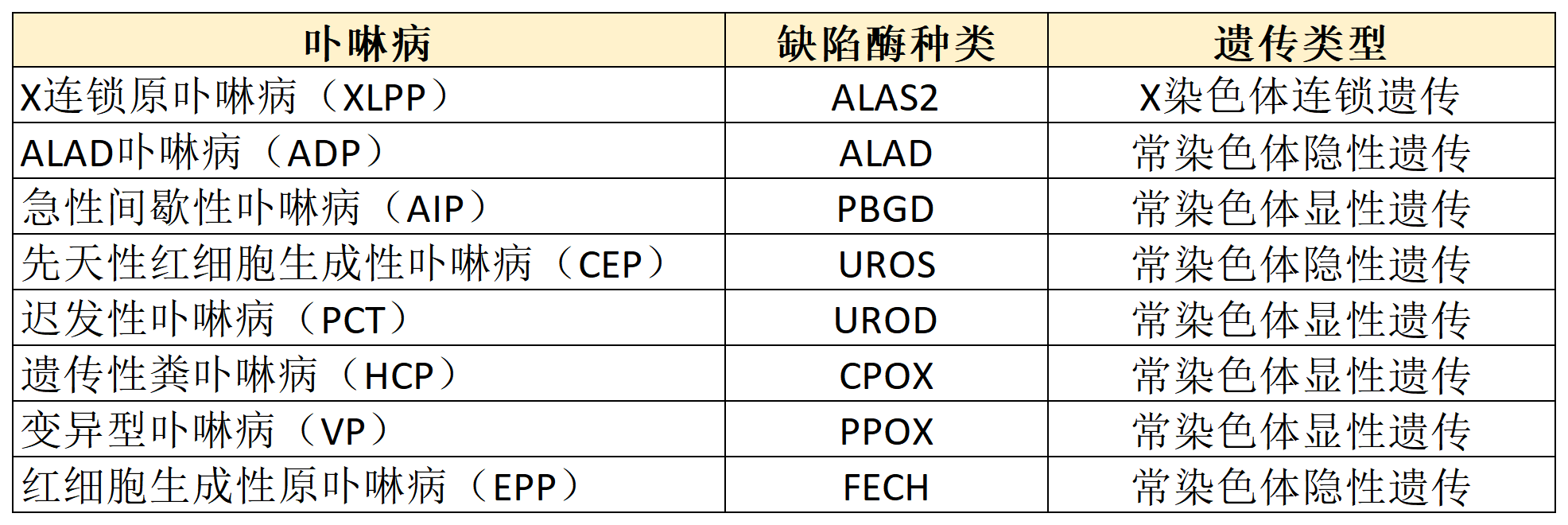
皮肤型卟啉病以PCT最常见,EPP、XLPP等较少见。其临床表现多样,主要为光敏性皮肤损害,如日光暴露部位慢性或急性皮损,可为水疱性或非水疱性,常伴有瘢痕形成和色素改变。神经症状型卟啉病以AIP最多见,主要表现为发作性神经精神性症状,如急性腹痛、周围神经病(如感觉减退、肌无力)、自主神经症状(如心悸、高血压、震颤)、精神症状(如焦虑、失眠、癔症)等,也可伴皮肤表现。混合型卟啉病较少见,患者同时存在皮肤表现及神经精神症状。而肝功能损伤可见于任一卟啉病类型,但以皮肤型卟啉病更常见,临床及病理表现多样,可为肝细胞损伤、胆管细胞受累、门脉周围纤维化、胆汁淤积或肝硬化等,肝脏病理可见特征性折光结晶。
卟啉病的诊断需要结合临床表现、血/尿液等检测、皮肤及肝脏组织病理学及基因检测。其中,血液、尿液、粪便中卟啉及其前体物质的定性或定量检测是初始诊断的常用方法,对区分卟啉病类型具有重要意义。血浆卟啉荧光分析可用于区分不同皮肤型卟啉病。基因突变检测则可以直接明确突变的基因及突变类型。肝脏影像学、组织学检查则可以通过有创及无创方式评估是否存在肝脏受累及受累程度,亦有助于判断卟啉病分型。
卟啉病的治疗以去除诱因及对症治疗为主。皮肤型卟啉病应注意进行光防护,促进皮肤对阳光耐受力、增加色素沉着;PCT患者可尝试服用小剂量羟氯喹等促进卟啉前体物质从肝脏排除,提高酶活性;肝移植可延缓部分卟啉病患者的肝损伤,但无法完全治愈,造血干细胞移植或可获得更好疗效(但不包括肝卟啉病患者,如AIP、VP、HCP、ADP)。AIP患者需注意避免暴饮暴食、特殊药物(巴比妥类、镇静剂)等诱发因素,急性发作期可输注高铁血红素并进行热量支持。卟啉病患者(尤其急性肝卟啉病患者)肝癌发生率高,建议定期随访,加强检测。

赵鸿
北京大学第一医院感染疾病科,教授、主任医师
中华医学会感染病学分会常务委员、副秘书长
北京医学会感染病学分会委员
中国研究型医院学会感染病专业委员会 常务委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
●病例报告:首次报告威尔逊病合并双侧色素性视网膜炎(Front Med (Lausanne),2022, IF=5.085; Q3区)

●中国威尔逊病患者潜在修饰基因的鉴定(Metallomics,2022, IF=4.636; Q2区)
●葡萄牙北部威尔逊病:一项长期随访研究(Orphanet J Rare Dis,2022, IF=4.303; Q3区)
●用于诊断威尔逊病的计算参数(Singapore Med J,2022, IF=3.331; Q3区)
●卟啉症患者发生肝细胞癌的风险:系统评价(Cancers (Basel),2022, IF=6.575; Q2区)
●Givosiran 治疗成人急性肝卟啉症(Drug Des Devel Ther,2022, IF=4.319; Q2区)
●急性间歇性卟啉病:中国致病性 HMBS 变异的流行情况及中国河北省的流行病学调查(Ann Transl Med,2022, IF=3.616; Q2区)
●放血在红细胞生成性原卟啉症相关肝损害治疗中的作用(Sci Rep,2022, IF=4.996; Q1区)
●靶向代谢组学评估急性间歇性卟啉症患者的代谢变化(Int J Mol Sci,2022, IF=6.208; Q2区)
●急性间歇性卟啉症治疗新策略(Biomedicines,2022, IF=4.757; Q2区)
三、临床资讯
1 幻灯分享
1.1标题及链接:脾大贫血皮疹待查
1.2 病例分享
患者女,43岁,因“发现脾大13年,加重并皮疹3年”入院。
病史:患者2003年体检发现脾脏增大(4×300px),2013年无诱因间断出现全身丘疹伴瘙痒、轻微疼痛,无水疱、破溃,可自行消退,部分残留色素沉着。此后3年内多次查血示肝功能异常、血细胞减少,间断口服熊去氧胆酸等症状仍反复。2016-07查腹部超声示肝硬化、脾大、腹水、门脉高压。2月内体重下降3.5Kg,自幼紫外线照射后皮肤出现皮疹,否认关节痛、晨僵、口干等,否认长期或特殊药物服用史,否认过敏史。育有1女体健,母亲、哥哥均有“哮喘”,余家族史无特殊。
入院查体:T 36.6℃ P 99次/分 R 12次/分 BP 108/69mmHg。全身皮肤黏膜黄染,可见散在分布的丘疹、色素沉着,以手背和上肢、面部为主。浅表淋巴结多处肿大、无触痛、直径不足25px。肝肋下125px,质韧表面不光滑,无触痛;脾肋下150px。
辅助检查:WBC 3.20×10^9/L,HGB 79g/L,PLT 37×10^9/L,Ret 74.20×10^9/L;ALT 156IU/L,AST 268IU/,ALB 36.1g/L,ALP 105IU/L,GGT 217IU/L,TBil 79.3μmol/L,Scr 71.00 μmol/L,NH3 83.0↑μmol/L;PTA 51%;ESR 33mm/第1小时末。HBV DNA、HCV RNA、CMV /EBV DNA(血清及淋巴细胞)(-)。抗骨骼肌抗体1:100阳性。 IgG 19.00g/L,IgA 5.06g/L,IgM 2.28 g/L,C3 0.758 g/L,C4 0.115 g/L,ASO 50.60 IU/mL,CRP 10.30mg/L,铜蓝蛋白 243.00 mg/L。
骨髓穿刺:骨髓增生,以巨核系及红系增生为主,可见噬血细胞。
PET-CT :惰性血液肿瘤可能大(Castleman’s disease?):肝脏肿大,形态不规则,葡萄糖代谢不均匀增高;胃周、肝门区、腹主动脉旁多发肿大淋巴结;脾脏明显增大,胸、腹、盆腔多发积液,葡萄糖代谢均未见增高。
皮肤活检:表皮轻度乳头瘤样增生,真皮浅层散在噬黑素细胞,真皮浅中层血管周围灶状淋巴细胞浸润。
肝脏活检(2016-08-02):肝细胞、扩张的毛细胆管、肝窦Kupffer细胞及汇管区内可见棕褐色大小不等的团块状或颗粒样沉积物偏振光下见Maltese十字结构;汇管区扩大、纤维组织沉积、细胆管反应增生呈枝芽状;CK-7免疫染色示较多肝细胞呈阳性反应。
尿液:尿卟胆原-、尿卟啉-。
红细胞内锌卟啉62.1ug/gHb(0-4.7)。
血浆荧光发射峰检测:峰值634nm左右。
基因检测:c1706-1709 del. AGTG(患者),其母亲、弟弟、女儿均(-)。
诊断:X -连锁原卟啉病(XLPP),肝硬化失代偿期、门静脉高压、食管胃底静脉曲张、脾大 脾功能亢进,腹腔积液。
治疗:予精氨酸血红素、碳水化合物负荷(Glu 300g/d),并对症保肝(熊去氧胆酸、还原性谷胱甘肽)、治疗三系减低(巨和粒、速力菲等),辅以新鲜冰冻血浆支持治疗。
随访:2月后复查病情稳定,ALT 11IU/L,GGT 179IU/L,HGB 94g/L。
讨论:X连锁原卟啉病是红细胞特异性ALAS2基因变异引起的卟啉代谢障碍性疾病。ALAS2活性升高,使红细胞内游离原卟啉Ⅸ及锌-原卟啉Ⅸ升高,原卟啉生成与代谢失衡,引起皮肤、肝脏等组织器官受累。临床表现常见非水疱性痛性皮疹、肝功能异常,重者可出现肝硬化、门脉高压。
因此提高对卟啉症临床特征的认识至关重要,尤其在不明原因肝硬化患者,胆汁淤积性肝病伴皮肤病变可能是卟啉病的第一线索,对于无条件行肝穿刺活检的患者如失代偿期肝硬化者,应行尿液、粪便、红细胞内游离卟啉及其代谢产物检测,血浆荧光发射峰检测及基因检测明确诊断。
1.3供稿专家简介
赵鸿
北京大学第一医院感染疾病科,教授、主任医师
中华医学会感染病学分会常务委员、副秘书长
北京医学会感染病学分会委员
中国研究型医院学会感染病专业委员会 常务委员
四、联系方式
●投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
●联系电话:010-63291007
●联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间