
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张敏
执行编辑:郑素军,梁晨,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
骨耳肝肠综合征(osteo-oto-hepato-enteric syndrome,O2HES)表现为胆汁淤积症、先天性腹泻、听力受损和骨骼脆弱四个临床表型的随机组合,2018年Alexandre Fabre等通过基因筛选和体外功能研究确定了UNC-45肌球蛋白伴侣A (UNC45A)基因突变或缺失是引起O2HE综合征的原因,其遗传规律符合常染色体隐性遗传。UNC45A属于UCS蛋白家族(UNC-45/CRO1/She4p),在各组织中广泛表达,参与细胞分裂或胞吐等细胞骨架功能。UNC45A缺失如何引起O2HES的机制尚不明确,但近期研究在体外肝细胞和肠上皮细胞中使用基因编辑和定点突变的方法证明,UNC45A是一种肌球蛋白(共)伴侣蛋白,其功能缺失可能降低肌球蛋白Vb(MYO5B)的表达,而MYO5B功能缺失可引起进行性家族性肝内胆汁淤积6型(PFIC6),以先天性腹泻和肝内胆汁淤积症状为特征。关于UNC45A突变导致O2HES更加深入的发病机制研究还有待进一步研究。作为一种新鉴别的疾病,O2HE综合征发病率尚不明确。
O2HES的临床表现多变,包括骨耳肝肠等四个方面的症状,根据UNC45A基因突变的不同将不同症候群组装在一起。骨骼方面可表现为骨脆伴复发性骨折、先天性关节发育不良、股骨头坏死、矮小等;耳方面可有知觉性耳聋、听力下降等;肝方面出现先天性复发性胆汁淤积、肝纤维化、肝衰竭,GGT可正常或升高;肠方面可有顽固性腹泻,甚至需要肠外营养。部分患者还存在轻度生长发育延迟、智力轻度低下等。其临床症状变异大、轻重不一,作为新识别的一种罕见病,临床医师尚缺乏认识,易导致漏诊、延迟诊断,误诊。本期月报从肝病科医生角度报道1例以慢性胆汁淤积反复发作、肝纤维化、轻度听力下降为主要表现的病例,该病例自出生起反复胆汁淤积,伴皮肤轻度瘙痒,可自行缓解,但肝纤维化、脾大,原因不明。经过肝组织病理学及相关基因检测后最终诊断明确。通过对该病例的介绍和展示,期望临床医生认识该病,对于原因不明的胆汁淤积,积极进行相关基因检测,警惕遗传代谢疾病等罕见病存在。

张敏
医学博士,主任医师,教授,硕士生导师
解放军总医院第五医学中心肝病科三病区主任
中华医学会肝病学分会遗传代谢性疾病学组委员
中华医学会医学遗传学分会生化和代谢学组委员
北京医学会遗传代谢病分会委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.病例报告:一例中国新生儿UNC45A相关骨耳肝肠综合征(European journal of medical genetics,2022, IF=2.565; Q3区)

2.胆盐输出泵缺陷症的基因型-表型关系(JHEP Rep,2023, IF=9.917; Q1区)
3.儿童进行性家族性肝内胆汁淤积症3型(PFIC3)患者的临床和遗传特征分析(Orphanet journal of rare diseases,2022, IF=4.303; Q2区)
4.Maralixibat减轻胆盐输出泵缺陷症患者的瘙痒程度的生物标志物分析(Metabolites,2022, IF=5.581; Q2区)
5.Odevixibat治疗进行性家族性肝内胆汁淤积的III期试验(The lancet,2022, IF=45.042; Q1区)
6.肝移植治疗儿童进行性家族性肝内胆汁淤积症的长期结局(Journal of clinical medicine,2022, IF=4.964; Q2区)
7.ABCB4、ABCB11和ATP8B1变异患者成人发病的临床表型(Hepatology communications,2022, IF=5.701; Q2区)
8.综述:获得性和遗传性胆汁淤积症的基因治疗(Biomedicines,2022, IF=4.757; Q2区)
9.综述:进展性家族性肝内胆汁淤积症(Clinics in liver disease,2022, IF=6.265; Q2区)
10.病例报告:进展性家族性肝内胆汁淤积症3型:中国ABCB4变异特征(Frontiers in medicine,2022, IF=5.058; Q2区)
三、临床资讯
1.1病例分享
患者男性,18岁,因间断身黄、眼黄18年,伴皮肤瘙痒入院。
现病史:患儿于2004年10月出生后眼黄、尿黄,当地医院检查肝功能异常(具体不详),经中成药物治疗后可正常。此后2年内约出现3次眼黄、尿黄伴皮肤瘙痒,先后在当地医院住院,查转氨酶及胆红素水平均高(具体不详),HBVM、抗HCV、EBV、CMV病毒学标志物均阴性,予以保肝等治疗。2007年10月当地医院查ALT<25U/L,TBIL 146 µmol/L,DBil 42µmol/L,腹部B超提示脾脏稍大,按巨细胞病毒感染应用更昔洛韦、干扰素抗病毒治疗,同时予以保肝、降酶等治疗,肝功仍间断异常。2007年11月来我院就诊,TBil 22.3µmol/L,DBil 14.7µmol/L,ALT 116U/L,AST 130U/L,GGT166U/L,铜蓝蛋白 0.57g/L。行肝穿检查,病理:肝硬化活动性并轻度局灶性肝细胞淤积。2010年8月,患者因肝功仍轻度异常,住院行二次肝穿病理检查提示早期肝硬化,病变轻微活动,考虑慢性非嗜肝病毒感染。此后院外定期复查肝功,ALT、AST波动于20-50U/L之间,多次出现黄疸,均可自行恢复正常。2022年8月下旬患者再次出现皮肤瘙痒,夜间重,伴皮肤黄染,无发热、腹泻、咳嗽、咳痰、皮疹等症状,加用熊去氧胆酸治疗,9月8日自觉皮肤瘙痒及黄染症状进行性加重,当地化验:TBil 118µmol/L,DBil 93µmol/L, ALT 31U/L,AST 37U/L,ALP 234U/L,GGT 26U/L,为进一步诊治来我院,2022-8门诊以“黄疸”收入我科。
既往史及个人史:生于原籍,第2胎,足月顺产,出生时体重3.5Kg,母乳喂养。2010年12月因“双眼白内障”行手术治疗。否认伤寒、结核等传染病史,否认慢性病史,否认其他外伤及手术史,否认药物过敏史,按时进行预防接种。
家族史:父母体健,为姨表亲结婚,否认遗传性疾病史。
入院查体:身高:4200px,体重:55Kg,神志清,精神可,记忆力、定向力、计算力均正常,肝掌阴性,未见蜘蛛痣,皮肤及巩膜重度黄染,心肺腹查体无异常,双下肢无水肿,生理反射存在,病理征未引出,扑翼样震颤阴性。
入院后完善检查:血常规:WBC 7.08×109/L,N 63.2%,RBC 4.29×1012/L,HGB 140g/L,PLT 278×109/L。肝功:ALB 40g/L,TBil 171.2µmol/L,DBil 139.4µmol/L,ALT 31U/L,AST 44U/L,ALP 262U/L,GGT 25U/L,BA 292µmol/L,肾功能、电解质正常,甲、乙、丙、戊病毒标记物均阴性。CMV及EBV IgG、IgM、DNA均阴性。铜蓝蛋白 0.65g/L。自身抗体系列均阴性。免疫球蛋白水平均正常。淋巴细胞亚群水平正常。
腹部B超结果:肝回声增粗(肝损害结合临床)、脾大。
腹部MRCP结果:影像所见:肝内胆管及胆总管未见扩张。胰管未见扩张。
无创肝结果:肝脏硬度值(Stiffness):18.8 Kpa;脂肪衰减:188(db/m)
肝活组织病理检果:
●肝硬化活动性并轻度局灶性肝细胞淤积(2007-11)。
●早期肝硬化,病变轻微活动,考虑慢性非嗜肝病毒感染(2010-04)。
基因检测结果:在 UNC45A 基因发现一处纯合变异。c.292C>T chr15:91479556 p.Arg98Trp。NM_018671; exon4。

一代测序结果
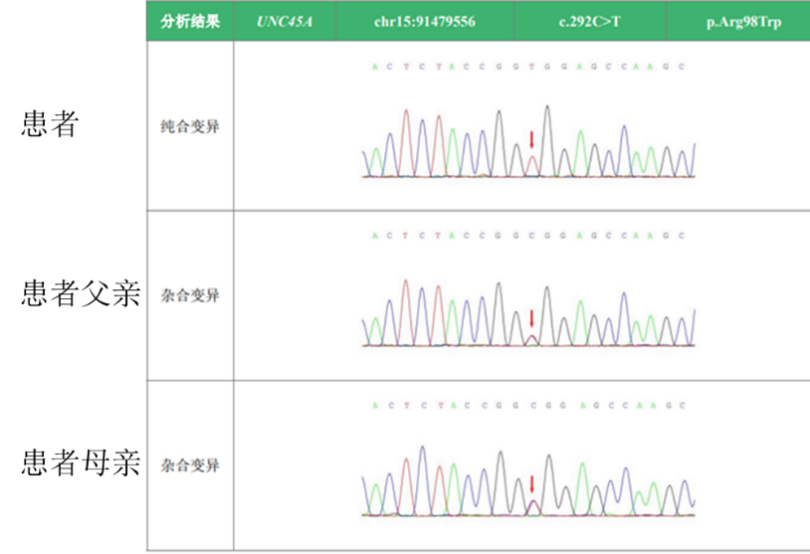
综上,患者临床表现为反复发生肝内胆汁淤积伴皮肤瘙痒,伴转氨酶升高、脾大。肝脏活组织病理检查显示:肝硬化活动性、肝内胆汁淤积。基因检测该样本在 UNC45A 基因发现一处纯合变异,c.292C>T。患者从婴儿期至今反复出现肝功能波动,予以保肝、降酶、退黄等治疗后或不予治疗情况下肝功能可恢复至正常水平,可除外病毒性肝病、自身免疫性肝病、肝豆状核变性、脂肪性肝病、药物及毒物肝损伤等病因,结合患者既往病史、父母近亲结婚特殊家族史及基因检查结果,经查阅文献,考虑该患者诊断为骨-耳-肝-肠综合征可能。该病表型可在四个系统的症状中随机组合呈现。该患者有肝病表现,基因结果回报后仔细反复追问病史,有无骨、耳、肠方面的症状体征。患者诉幼年时有左手腕和小指骨折史,已痊愈未告知医生,其它部位骨折不明确;听力似略有减退,但对日常交流影响不大,在医生建议下近期到专科行听力检测,发现双耳存在轻-中度感音性耳聋。无腹泻,未见其它明确肠道症状,有待随访观察及进一步专科检查。
治疗:应用熊去氧胆酸、复方甘草酸苷、丁二磺酸腺苷蛋氨酸、考来烯胺等对症处理,肝功可复常,但似乎与治疗无明显相关。目前患者仍在随访中。
1.2供稿专家简介
张敏
医学博士,主任医师,教授,硕士生导师
解放军总医院第五医学中心肝病科三病区主任
中华医学会肝病学分会遗传代谢性疾病学组委员
中华医学会医学遗传学分会生化和代谢学组委员
北京医学会遗传代谢病分会委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间