
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:侯维
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
钠牛磺胆酸共转运多肽(Sodium-taurocholate cotransporting polypeptide,NTCP)缺陷病是一种SLC10A1双等位基因变异引起的胆汁酸代谢障碍性疾病,为常染色体隐性遗传。SLC10A1基因定位于染色体14q24.2,全长23kb,包含5个外显子、4个内含子,编码的NTCP是一种主要表达在肝细胞基底外侧膜的多跨膜糖蛋白,以钠依赖的方式将大部分结合胆汁酸从门静脉摄取入肝细胞,维持胆汁酸肠肝循环稳态。除此,NTCP可以介导甲状腺素、类固醇激素、药物等转运,还是HBV、HDV进入肝细胞的功能性受体。
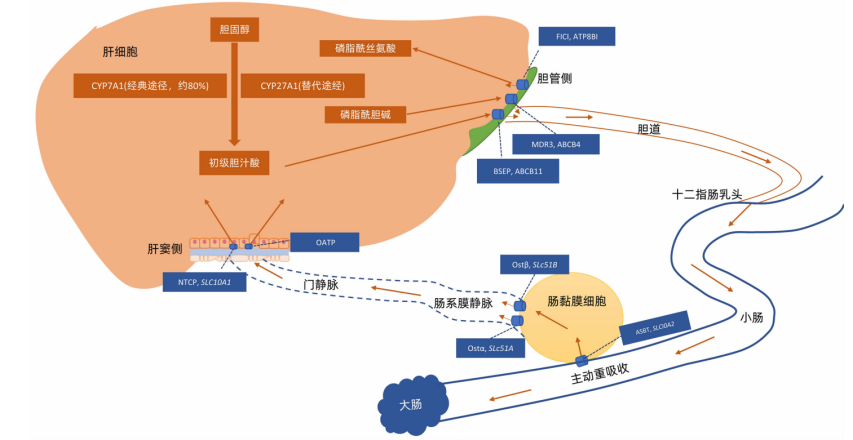
图1 胆汁酸代谢过程[1]
SLC10A1变异等位基因频率具有种族差异,根据基因组聚合数据库 (gnomAD),c.800C>T(p.Ser267Phe)在东亚人种的等位基因频率可高达0.046,而在欧洲及中东人中则为阴性。尽管该变异在人群中频率较高,但体外功能学研究证实c.800C>T(p.Ser267Phe)会导致NTCP摄取胆汁酸的功能几乎完全丧失[2]。SLC10A1基因c.800C>T(p.Ser267Phe)纯合变异是我国NTCP患者中最常见的基因型。广东省NTCP缺陷病流行病学数据显示,SLC10A1变异等位基因频率为11.72% (663/5656),NTCP 缺陷病理论患病率为 1.37%[3]。 既往国内外报道NTCP缺陷病患者的SLC10A1基因变异位点还包括:c.263T>C (p.Ile88Thr)、c.595A>C (p.Ser199Arg)、c.615_618delCTCT (p.S206Pfs*12)、c.665T>C (p.Leu222Ser) 、c.745C>T (p.R249W)、c.755G>A (p.Arg252His)等。
NTCP缺陷病主要的临床表现为血清总胆汁酸水平升高,与其他肝功指标变化不同步。可伴有25羟维生素D缺乏,胆囊息肉及胆结石发生率增高[4-5]。儿童患者胆汁酸水平显著升高,可以伴随高胆红素血症,考虑其原因为:作为NTCP缺陷病的代偿机制,肝细胞基底侧二聚体OATP1B1/1B3能够以非钠依赖方式摄取胆汁酸、胆红素,但该二聚体发育成熟具有明显年龄依赖性,NTCP缺陷病患儿的高胆汁酸水平可随年龄增长而改善[6]。成人患者多缺乏阳性症状与体征,仅检测发现血清总胆汁酸轻微升高,部分女性患者被诊断为妊娠期肝内胆汁淤积(Intrahepatic cholestasis of pregnancy,ICP)。
NTCP缺陷病尚缺乏临床和生化诊断标准,肝脏组织特征缺乏特异性,可见肝细胞肿胀,轻微肝细胞内淤胆、汇管区轻度炎细胞浸润[7]。SLC10A1基因检测发现双等位基因致病性变异是可靠的确诊依据。NTCP缺陷病缺乏特异性治疗药物,对症支持治疗是主要的管理手段。目前文献报道的NTCP缺陷病患者短期临床结局均良好,无肝硬化等严重预后的报道。NTCP缺陷病对妊娠期的影响尚不明确,既往报道妊娠合并NTCP缺陷病3例,分别于妊娠38周胎膜早破胎粪污染导致死胎、妊娠38周顺产活婴、妊娠34周剖宫产双胎,三位患者产后血清胆汁酸水平均不能降至正常。既往研究还发现妊娠期高胆汁酸血症与胎儿出生低体重和生长受限风险增加有关[8]。对NTCP患者妊娠期胆汁酸谱的研究可能有助于深入探索NTCP与ICP的相关性。
本期月报报道1例成人SLC10A1变异的NTCP缺陷病患者,该患者因间断高胆汁酸血症、影响正常妊娠而就诊我院寻找病因,最终经过基因检测明确诊断。由于SLC10A1变异位点在正常人群中等位基因频率较高,在全外显子检测的数据分析过程中可能被划分为单核苷酸多态性而被遗漏。因此,提高临床医生对该疾病的认识,增强临床医师与检测机构之间的沟通,有助于部分高胆汁酸血症患者明确诊断。此外,目前主要研究认为NTCP缺陷病为一良性过程,预后良好,加深对此病的认识和理解有助于减轻患者对此病不必要的焦虑,避免不必要的检查和干预。
参考文献
1. 易波,李雪,汤善宏.基因突变致胆汁酸代谢异常相关发生机制的研究进展[J].临床肝胆病杂志,2022,38(9):2136-2140.
2.Ho RH, Leake BF, Roberts RL, et.Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem,2004,279(8):7213-7222.
3.林桂枝.广东人群钠牛磺胆酸共转运多肽缺陷病分子流行病学调查[D].广东:暨南大学,2020.
4.Liu R, Chen C, Xia X, Liao Q, et al.Homozygous p.Ser267Phe in SLC10A1 is associated with a new type of hypercholanemia and implications for personalized medicine. Sci Rep,2017,7(1):9214.
5.Mao F, Wang MX, Hou X, et al.NTCP Deficiency Causes Gallbladder Abnormalities in Mice and Human Beings. Cell Mol Gastroenterol Hepatol,2021,11(3):831-839.
6.宋元宗.钠牛磺胆酸共转运多肽缺陷病的发病机制、临床表现及诊疗进展[J].临床肝胆病杂志,2019,35(8):1690-1692.
7.Dong C, Zhang BP, Wang H, et al.Clinical and histopathologic features of sodium taurocholate cotransporting polypeptide deficiency in pediatric patients. Medicine (Baltimore),2019,98(39):e17305.
8.Song F, Chen Y, Chen L, et al. Association of Elevated Maternal Serum Total Bile Acids With Low Birth Weight and Intrauterine Fetal Growth Restriction. JAMA Netw Open,2021,4(7):e2117409.

侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.有机阴离子转运多肽 (OATP) 1B3 是人类肝脏摄取结合胆汁酸的重要转运蛋白(Cellular and molecular gastroenterology and hepatology, 2023,IF=7.20; Q1区)

2.Maralixibat 治疗进行性家族性肝内胆汁淤积症 (MARCH-PFIC):一项多中心、随机、双盲、安慰剂对照的 3 期试验。(The lancet. Gastroenterology & hepatology, 2024,IF=35.70; Q1区)
3.基因型与紧密连接蛋白2(TJP2)缺乏型胆汁淤积患儿的临床病程和结局相关(Hepatology, 2024,IF=13.50; Q1区)
4.四硫代钼酸盐对健康志愿者和Wilson病患者铜代谢的影响。(Journal of hepatology, 2024,IF=25.70; Q1区)
5.青霉胺与锌联合治疗的肝性Wilson病患者停用青霉胺后病情复发研究(Journal of pediatric gastroenterology and nutrition, 2024,IF=2.90; Q2区)
6.WilsonGenAI:一种对Wilson病致病变异进行分类的深度学习方法(PloS one, 2024,IF=3.70; Q2区)
7.Crigler-Najjar综合症:展望未来并不会让我们忘记现在(Orphanet journal of rare diseases, 2024,IF=3.70; Q2区)
8.综述:妊娠期肝内胆汁淤积症的发病机制和组学研究进展(Hepatology international, 2024,IF=6.60; Q1区)
9.综述:儿科研究为儿童和成人胆汁淤积症的治疗设定了新标准(The Lancet. Child & adolescent health, 2024,IF=36.40; Q1区)
10.病例报道:钠牛磺胆酸共转运多肽缺陷病的两对双胞胎(Clinics and research in hepatology and gastroenterology, 2024,IF=2.70; Q3区)
三、临床资讯
3.1 病例分享:1例成人NTCP缺陷病
患者女性,30岁,因“间断血清总胆汁酸升高12年”入我院肝病中心一科。
现病史:患者12年前于妊娠37周时因皮肤瘙痒就诊,无恶心、呕吐、厌油腻、皮肤巩膜黄染等不适,化验发现总胆汁酸>100μmol/L,转氨酶正常(具体不详),行剖腹产后未进一步诊治。4月前患者于妊娠6周时出现胎停,伴皮肤瘙痒,就诊于当地医院化验提示总胆汁酸38.42μmol/L,余肝功能正常,接受清宫术。2月前患者在当地医院复查:ALT 88.6U/L,AST 50.4U/L,GGT 48.2U/L,ALP 113.5U/L,TBIL 31.5μmol/L,DBIL 9.73μmol/L,IBIL 21.77μmol/L,TBA 70.2μmol/L,血常规未见异常。上腹增强MRI:肝脏、胆囊、胰腺及双肾未见占位征象,脾大。MRCP未见异常,开始口服“熊去氧胆酸、丁二磺酸腺苷蛋氨酸片”治疗,患者自述胆汁酸升高未缓解。现为明确病因,收入我院。
既往史及家族史:平素健康状况良好,否认服药史,无饮酒史。父亲患慢性乙型病毒性肝炎,母亲体健。
查体:体温36.9℃,血压114/67mmHg,BMI 18.32kg/m2,神志清楚,营养良好,皮肤巩膜无黄染,心肺查体未见异常,腹软,肝脾未触及,双下肢无水肿。
实验室检查:血常规:WBC 6.47×109/L,N% 62%,HGB 125g/L,PLT 229×109/L,网织红细胞绝对值 46.1×109/L;生化:ALT 13U/L,AST 20U/L,γ-GT 13U/L,ALP 59U/L,TBIL 19.2μmol/L,DBIL 6.2μmol/L,ALB 46.2g/L,TBA 82.8μmol/L,Cr 53μmol/L,GLU 4.43mmol/L,TG 0.83mmol/L,TC 5.53mmol/L,HDL-C 2.41mmol/L,LDL-C 2.36mmol/L;PTA 102%;AFP正常;乙肝、丙肝阴性;ANA1:100(核颗粒型),免疫球蛋白、自身抗体谱、抗核抗体谱、ANCA、AMA-M2未见异常;铜蓝蛋白正常;甲状腺功能未见异常;Coombs阴性;外周血红细胞形态大致正常。
影像学检查:肝胆胰脾彩超:未见异常。肝脏弹性测定:E 3.8kPa,CAP 195dB/m。
肝脏病理:肝穿刺组织肝小叶结构可辨,轻度肝细胞水样变性,少量肝细胞脂肪变性,少数肝细胞毛玻璃样变,个别点灶状坏死,偶见凋亡小体,窦周炎不显著;汇管区未见扩大,炎细胞不明显,无界面炎。结论:结合临床考虑轻度药物或环境类毒素诱导性病变,病变程度相当于G0-1S0。
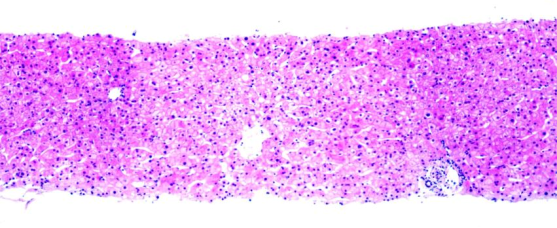
图1 肝活检显示轻度肝细胞水样变性,少量肝细胞脂肪变性,少数肝细胞毛玻璃样变,个别点灶状坏死,偶见凋亡小体。(HE染色,10×)
全外显子基因测序结果及家系验证:
1.SLC10A1基因c.800C>T(p.S267F),纯合变异,ACMG评级为意义不明(BS1+PS3+PM3+PP3+PP4),来源父母。
2.SLCO1B3基因c.508_509del(p.M170fs),杂合变异,ACMG评级为致病的(PVS1+PM2),来源父亲。
3.SLCO1B3基因c.251C>A(p.S112Y),杂合变异,ACMG评级为意义不明(PM1+PP3),来源父亲。
4.ATP8B1基因c.2534A>G(p.K845R),杂合变异,ACMG评级为意义不明(PM2_supporting+BP4)。
5.UGT1A1基因c.211G>A(p.G71R),杂合变异,ACMG评级为意义不明。
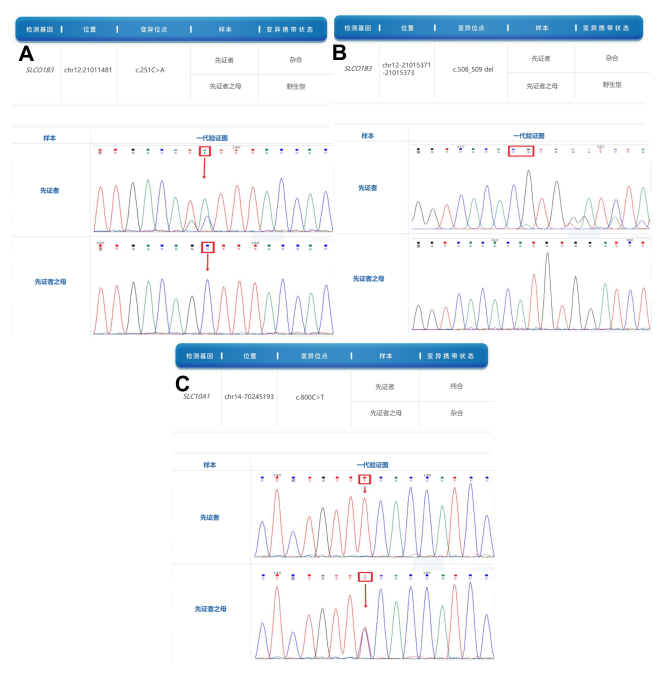
图2 患者基因变异的一代验证。A图示患者SLCO1B3基因c.251C>A(p.S112Y),杂合变异,来自父亲;B图示患者SLCO1B3基因c.508_509del(p.M170fs),杂合变异,来自父亲;C图示患者SLC10A1基因c.800C>T(p.S267F),纯合变异,来自父亲和母亲。
该患者虽检出了SLCO1B3的2个变异位点,但均来源于父亲,且未检出SLCO1B1基因变异,不支持Rotor综合征诊断。此外,检出的ATP8B1基因和UGT1A1基因均为单杂合变异,且与临床表型不符,也不支持相应的诊断。
诊断:根据患者间断血清总胆汁酸升高病史,SLC10A1基因c.800C>T(p.S267F)纯合变异,诊断为NTCP缺陷病。
随访:出院后患者短暂口服消胆胺治疗,后无不适症状遂停药。出院后5月检测血总胆汁酸22μmol/L,出院后7月检测总胆汁酸34.1μmol/L。
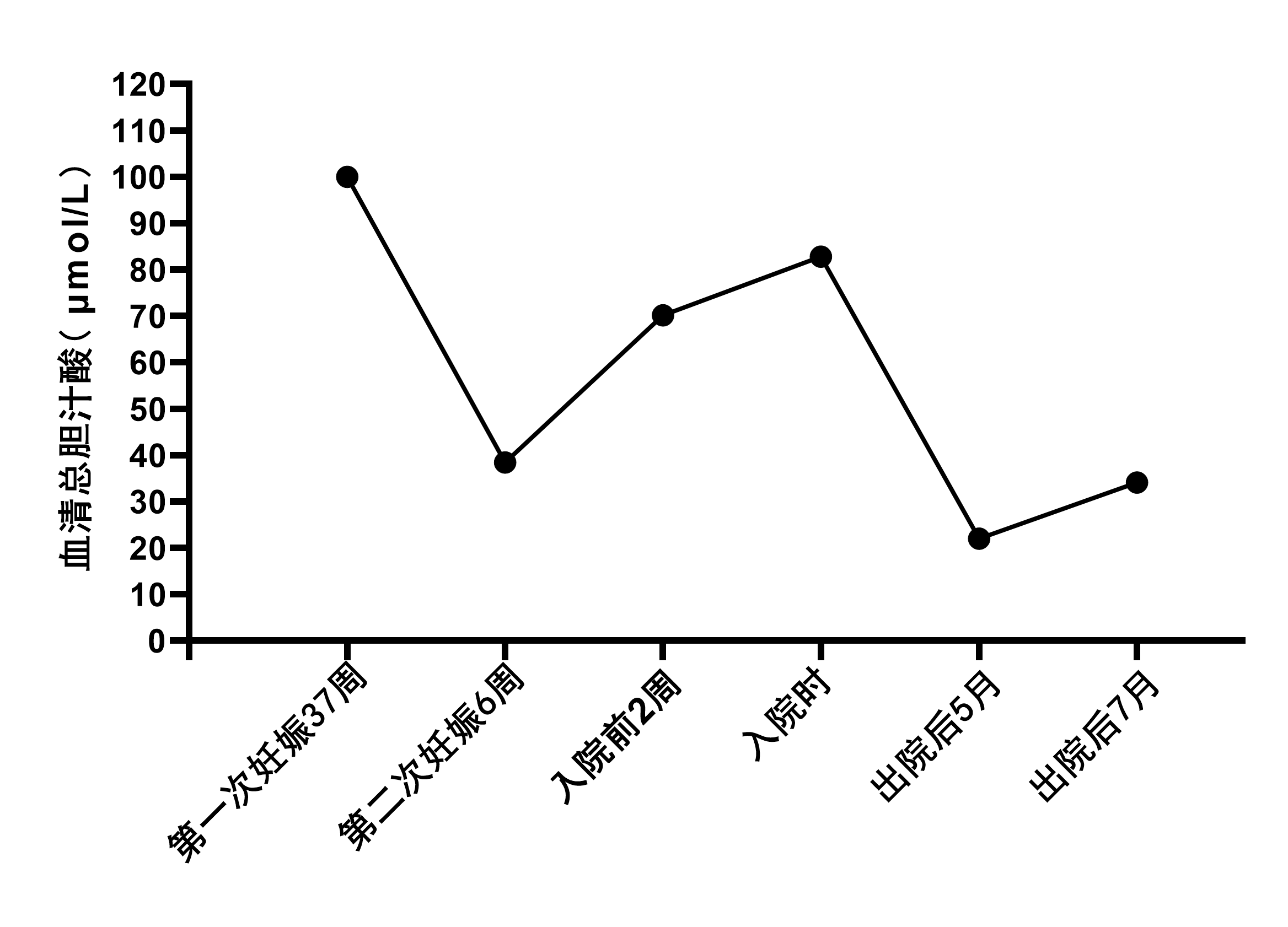
图3 患者血清总胆汁酸变化趋势
3.2 供稿专家简介
侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员

於海天
首都医科大学附属北京佑安医院内科学博士
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间