
本文转载自“佑安肝病感染病专科医疗联盟”
No.1遗传代谢性肝病月报
主管:佑安肝病感染病专科医疗联盟(YouMe AID)
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会
指导:段钟平
总编辑:郑素军
本期责任主编:郑素军
执行编辑:白洁,孔明
No.2本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、专业委员会名单
五、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
遗传代谢性肝病月报主要包括以下栏目,力求“一眼抓住最新进展,短时积累临床心得!”
“学术进展”:通过文献分享、专家点评,帮您快速了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座、学会动态等信息,助您感悟遗传代谢性肝病临床诊治之奥妙!
每月一报,让遗传代谢性肝病从罕见病、疑难病变为医者眼中的熟悉病、简单病!
每月一报,助力全面提高我国遗传代谢性肝病的诊治与科研水平!
尽管新型冠状病毒感染的肺炎疫情仍然笼罩神州大地,第一期遗传代谢性肝病月报仍然按时推出了!本期在归纳、介绍2019年12月份文献进展基础上,重点关注“肝豆状核变性(Wilson病)”等金属代谢障碍性疾病,分享了“肝豆状核变性”典型病例以及讲座幻灯。
最后,再次向奋战在抗击疫情一线的同行们致敬!您的健康平安就是专委会最大的心愿!

郑素军 主任医师
首都医科大学附属北京佑安医院疑难肝病及人工肝中心主任医师,教授、博士研究生导师。中华医学会肝病学分会遗传代谢性肝病协作组委员,秘书长。佑安专科联盟遗传代谢性肝病专业委员会主任委员。
邮箱:zhengsujun003@126.com。
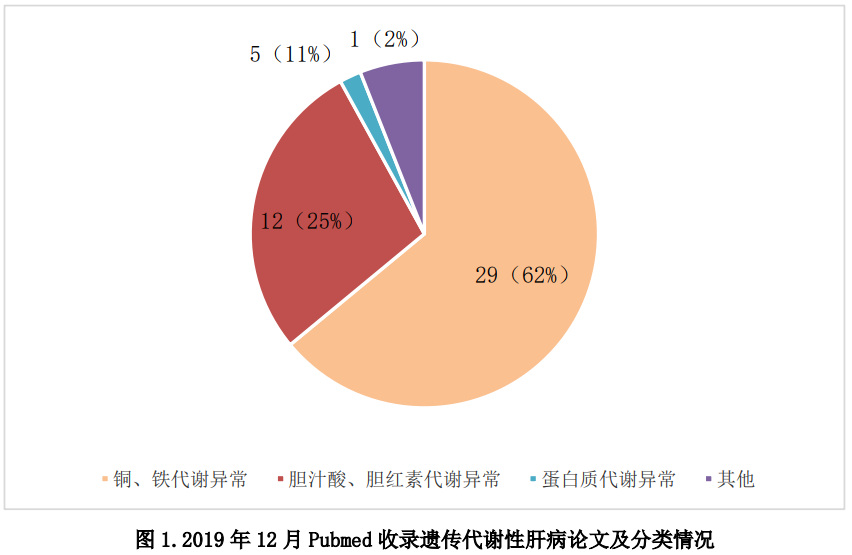
1.Pubmed收录论文归类
2019年12月发表遗传代谢性肝病相关论文共50篇,按遗传代谢性肝病常见分类(比如胆红素、胆汁酸、金属、蛋白、糖等代谢异常),统计结果如下图。
铁过载通过降低心脏线粒体功能、干扰线粒体动力学,引起心脏功能受损
(Arch Biochem Biophys, 2019, IF=3.559, Q2区)
点击下载:

高通量iPLEX基因分型检测方法可用于筛查希特林蛋白缺陷病(NICCD)引起的新生儿肝内胆汁淤积症
(J Inherit Metab Dis, 2019, IF=4.287, Q2区)
点击下载:
腺相关病毒介导的基因治疗成功治愈PFIC3模型小鼠
(Nat Commun, 2019, IF=11.878, Q1区)
点击下载:
基于质谱的蛋白质组学可鉴定肝豆状核变性小鼠模型肝脏分泌的血浆蛋白,为疾病进展的机制研究提供了新思路
(Metallomics, 2019, IF=3.571, Q2区)
点击下载:
美国研究表明,血色素沉着病占用大量医疗资源并造成巨大经济负担
(J Manag Care Spec Pharm, 2019, IF= 3.024, Q3区)
点击下载:
Ferroportin 1(FPN1)结构异常可导致血色素沉着病(4型)
(FASEB J , 2019, IF= 5.391, Q1区)
点击下载:
以AAV为载体的、截短的ATP7B基因治疗有望治愈肝豆状核变性
(Hum Gene Ther, 2019, IF=3.855, Q2区)
点击下载:
肝细胞癌合并或不合并血色素沉着病,肝移植/切除/射频消融/TACE治疗后,两组患者生存率无明显差别
(Am J Clin Oncol, 2019, IF=3.015, Q3区)
点击下载:
联合重组腺病毒-piggyBac转座酶的基因疗法可治愈PFIC 3型模型小鼠
(Hepatology, 2019, IF=14.971, Q1区)
点击下载:
铁螯合剂有望改善铁代谢异常引起的神经退行性疾病
(Brain Res Bull, 2019,IF=3.103, Q1区)
点击下载:
1
文献(英文):Tang N, SandahlTD, Ott P,et al.Computing the Pathogenicity of Wilson's Disease ATP7B Mutations: Implications for Disease Prevalence. J Chem Inf Model. 2019 Dec 23;59(12):5230-5243. Impact factor= 3.966, Q1。
文献(中文):Tang N, Sandahl TD, Ott P等。计算Wilson病ATP7B突变的致病性:对疾病患病率的影响.化学信息和建模杂志,2019,59(12):5230-5243. 影响因子= 3.966, Q1区。
英文摘要:
Genetic variations in the gene encoding the copper-transport protein ATP7B are the primary cause of Wilson's disease. Controversially, clinical prevalence seems much smaller than the prevalence estimated by genetic screening tools, causing fear that many people are undiagnosed, although early diagnosis and treatment is essential. To address this issue, we benchmarked 16 state-of-the-art computational disease-prediction methods against established data of missense ATP7B mutations. Our results show that the quality of the methods varies widely. We show the importance of optimizing the threshold of the methods used to distinguish pathogenic from nonpathogenic mutations against data of clinically confirmed pathogenic and nonpathogenic mutations. We find that most methods use thresholds that predict too many ATP7B mutations to be pathogenic. Thus, our findings explain the current controversy on Wilson's disease prevalence because meta-analysis and text search methods include many computational estimates that lead to higher disease prevalence than clinically observed. As proteins and diseases differ widely, a one-size-fits-all threshold cannot distinguish pathogenic and nonpathogenic mutations efficiently, as shown here. We also show that amino acid changes with small evolutionary substitution probability, mainly due to amino acid volume, are more associated with the disease, implying a pathological effect on the conformational state of the protein, which could affect copper transport or adenosine triphosphate recognition and hydrolysis. These findings may be a first step toward a more quantitative genotype-phenotype relationship of Wilson's disease.
中文摘要:
编码铜转运蛋白ATP7B的基因变异是Wilson病的主要病因。有争议的是,临床患病率似乎远远低于遗传筛查工具估计的患病率。(考虑到)早期诊断和治疗的必要性,这导致人们担心许多人被漏诊。为解决此问题,我们针对已建立的ATP7B错义突变数据,对16种最先进的(计算机)疾病预测方法进行了基准测试。我们的结果表明,这些预测方法的质量差异很大。优化预测方法的阈值,对于鉴别经临床确认的致病性和非致病性突变,有着重要作用-大多数方法若采用方法本身设立的默认阈值来预测,则使很多ATP7B突变会被判断为有致病性。因此,我们的发现解释了当前有关Wilson病的患病率方面的争议--目前的荟萃分析和文本检索中患病率的产生方法,包括了许多由计算机预测而产生的估计值,这导致疾病患病率高于临床上实际观察到的结果。由本研究可见,由于蛋白质和疾病的差异很大,因此“一刀切”的(软件)预测阈值无法有效地区分致病性和非致病性突变。我们还发现,进化替代概率较小的氨基酸发生变化(主要因氨基酸体积改变),其与疾病的关系更加密切,这暗示了其会对蛋白构象有不利影响,可能影响铜转运或三磷酸腺苷的识别和水解。这些发现可能是迈向(研究)Wilson病基因型-表型间定量关系的第一步。
张缭云教授简评
Wilson病是临床上常见的单基因、隐性遗传性疾病,但是其真正的流行情况,即患病率仍存在争议。既往报道该病的患病率约1/30000。近来,一些群体遗传学研究显示,肝豆状核变性的患病率显示高达1:7,026-1:4,000,差别如此之大,是研究方法的差异所致?还是的确有大量的患者未得到及时的诊治?
本项来自丹麦的研究给揭示上述真相提供了很好的研究思路。作者发现,最近的群体遗传学研究,对于新发现的突变基因的致病性,多是采用计算机软件预测,很多并没有经过功能验证。由于软件判断是否有致病性的阈值为默认设置,并不具有ATP7B蛋白或Wilson病特异性,软件预测为“有致病性”的突变,很可能是“良性突变”,并没有致病性。针对这一假设,作者采用WD 数据库中 (http://www.wilsondisease.med.ualberta.ca),根据突变致病性是否经临床证实,分为致病性突变和非致病性突变,对目前最常用的16种预测软件的预测准确性进行了检测,并对判断阈值进行优化,发现经优化的预测阈值能更准确地甄别致病性突变,由此来估算患病率,可能会更加接近临床实际。
另一方面,肝豆状核变性ATP7B基因型与表型间的关系,也远未阐明。本文中作者应用生物信息学分析,发现ATP7B基因突变导致的化学特征改变(例如被埋残基的侧链体积),是决定突变是否有致病性的重要因素,这种“蛋白结构-功能研究”可能有助于揭示基因型和表型间关系。
本研究优点:
1.思路巧:利用经临床证实的致病性和非致病性突变,对常见预测软件判断突变位点是否具有致病性的cut-off值进行了优化,提高了诊断Wilson病的准确性,也有助于揭示更为准确的Wilson病的患病率;
2.利用计算机软件等生物信息学分析方法,有助于理解突变是否具有致病性的相关机制。
本研究不足:针对Wilson病,在判断突变基因是否具有致病性方面,本研究虽然优化了预测软件的用于判断的Cut-off值或阈值,但没有利用临床队列对之进行验证。
可供借鉴或有所启迪之处:可借鉴本研究思路,对于Glibert/Crigler-najjar等其他遗传性疾病,在使用常用的计算机预测软件判断检出的基因突变是否有致病性时,其判断阈值也应当进行疾病特异性、个体化优化。
点击下载:
2
文献(英文):Wang CY, Xiao X, Bayer A, et al. Ablation of Hepatocyte Smad1, Smad5, and Smad8 Causes Severe Tissue Iron Loading and Liver Fibrosis in Mice. Hepatology, 2019Dec;70(6):1986-2002. Impact factor=14.971 , Q1.
文献(中文):Wang CY, Xiao X,Bayer A等.肝细胞Smad1,Smad5和Smad8缺陷导致小鼠的组织严重铁负荷和肝纤维化. 肝脏病杂志,2019Dec;70(6):1986-2002. 影响因子=14.971 , Q1区。
英文摘要:
A failure of iron to appropriately regulate liver hepcidin production is central to the pathogenesis of hereditary hemochromatosis. SMAD1/5 transcription factors, activated by bone morphogenetic protein (BMP) signaling, are major regulators of hepcidin production in response to iron; however, the role of SMAD8 and the contribution of SMADs to hepcidin production by other systemic cues remain uncertain. Here, we generated hepatocyte Smad8 single (Smad8fl/fl;Alb-Cre+ ), Smad1/5/8 triple (Smad158;Alb-Cre+ ), and littermate Smad1/5 double (Smad15;Alb-Cre+) knockout mice to investigate the role of SMAD8 in hepcidin and iron homeostasis regulation and liver injury. We found that Smad8; Alb-Cre+ mice exhibited no iron phenotype, whereas Smad158; Alb-Cre+ mice had greater iron overload than Smad15; Alb-Cre+ mice. In contrast to the sexual dimorphism reported for wild-type mice and other hemochromatosis models, hepcidin deficiency and extrahepatic iron loading were similarly severe in Smad15; Alb-Cre+ and Smad158; Alb-Cre+ female compared with male mice. Moreover, epidermal growth factor (EGF) failed to suppress hepcidin in Smad15; Alb-Cre+ hepatocytes. Conversely, hepcidin was still increased by lipopolysaccharide in Smad158; Alb-Cre+ mice, although lower basal hepcidin resulted in lower maximal hepcidin. Finally, unlike most mouse hemochromatosis models, Smad158; Alb-Cre+ developed liver injury and fibrosis at 8 weeks. Liver injury and fibrosis were prevented in Smad158; Alb-Cre+ mice by a low-iron diet and were minimal in iron-loaded Cre- mice. Conclusion: Hepatocyte Smad1/5/8 knockout mice are a model of hemochromatosis that encompasses liver injury and fibrosis seen in human disease. These mice reveal the redundant but critical role of SMAD8 in hepcidin and iron homeostasis regulation, establish a requirement for SMAD1/5/8 in hepcidin regulation by testosterone and EGF but not inflammation, and suggest a pathogenic role for both iron loading and SMAD1/5/8 deficiency in liver injury and fibrosis.
中文摘要:
肝铁调素产生异常是遗传性血色素沉着病的关键发病机制。由骨形态发生蛋白(BMP)信号激活的SMAD1/5转录因子是铁调节铁调素产生的主要因子。然而,SMAD8的作用以及SMAD通过其他系统对铁调素的调节作用仍不明确。我们构建了Smad8单株(Smad8fl/fl;Alb-Cre+),Smad1/5/8三株(Smad158;Alb-Cre+)和同窝出生的Smad1/5双株(Smad15;Alb-Cre+)敲除小鼠,以研究SMAD8在铁调素和铁稳态调节以及肝损伤中的作用。我们发现Smad8;Alb-Cre+小鼠没有铁负荷,而Smad158;Alb-Cre+小鼠比Smad15;Alb-Cre+小鼠具有更强的铁负荷。与野生型小鼠和其他血色素沉着病模型报道的两性差异相反,Smad15;Alb-Cre+和Smad158; Alb-Cre+的雌性小鼠和雄性小鼠的铁调素缺乏和肝外铁负荷同样严重。此外,表皮生长因子(EGF)不能抑制Smad15;Alb-Cre+小鼠肝细胞产生铁调素。相反,尽管较低的基础铁调素导致较低的最大铁调素,但在Smad158;Alb-Cre+小鼠中,脂多糖仍使铁调素增加。最后,与大多数小鼠血色素沉着病模型不同,Smad158; Alb-Cre+小鼠在8周时发展为肝损伤和纤维化。低铁饮食可预防Smad158;Alb-Cre+小鼠产生肝损伤和纤维化,而铁负载的Cre-小鼠产生肝损伤和纤维化很少。
结论:肝细胞Smad1/5/8基因敲除小鼠是一种血色素沉着症模型,涵盖人类疾病中可见到的肝损伤和纤维化。这些小鼠揭示了SMAD8在铁调素和铁稳态调节中的冗余但至关重要的作用,确定了SMAD1/5/8通过睾酮和EGF而不是通过炎症调节铁调素,并提出了铁负荷和SMAD1/5/8缺乏在肝损伤和纤维化中的致病作用。
肝铁调素(Hepcidin)产生异常是遗传性血色素沉着病的关键发病机制。骨形成蛋白(BMP)/Smad通路在铁调素调控中发挥了重要作用,有关SMAD1/5转录因子的作用较为清晰,但Smad8的作用尚不明确。该研究通过建立Smad8、Smad1/5/8、Smad1/5基因敲除小鼠模型,分别对铁调素产生相关影响调节因子进行了研究。发现Smad8在小鼠铁调素和铁稳态调节方面具有重要作用。肝细胞Smad1/5/8基因敲除小鼠可作为一种理想的血色素沉着症模型,体内铁负荷明显增加,且具备人类患者的肝损伤和纤维化表现。
铁调素是一种主要由肝脏合成的富含半胱氨酸、具有抗微生物活性的小分子阳离子多肽,也是机体铁稳态的主要调节激素。其编码基因位于19号染色体的长臂上(19q13),由3个外显子和2个内含子组成。该基因启动子区域含有多个顺式元件,能和CAAT增强子结合蛋白、核因子KB、肝细胞核因子(HNF)、缺氧诱导因子(HIF)、铁调素调节蛋白(HJV)和BMP/Smad等转录因子结合。铁调素通过抑制肠道铁吸收和网状内皮系统铁的释放维持铁稳态,是机体铁代谢的负调节因子。其主要在肝脏合成,在肾脏、心脏、骨骼肌、大脑、肺和胃中也有少量表达,小肠是铁吸收的主要器官,其上皮细胞有较高水平的表达。明确Smad8在铁调素调控中的作用对于进一步明确遗传性血色素沉着病的发病机制,寻求新的干预靶点具有重要参考价值。
点击下载:
4.本期Pubmed文献编译及短评专家简介
★ 文献编译 ★

白洁
首都医科大学附属北京佑安医院
在读博士研究生
佑安专科联盟遗传代谢性肝病专业委员会秘书
邮箱:docbai@yeah.net
★ 短评专家 ★

张缭云
主任医师,教授,硕士研究生导师
山西医科大学第一医院
感染病科主任
佑安专科联盟遗传代谢性肝病专业委员会副主任委员

李磊
主任医师,硕士研究生导师
中国科学技术大学附属第一医院
感染病科主任
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
三、临床资讯
1.典型病例分享
1 病例简介(肝豆状变性)
患儿李某某,男,11岁,主因“肝病史8年,腹胀1月”于2019-12-24收入院。
患者8年前体检时发现肝功能异常,谷丙转氨酶升高(具体不详)。无自觉不适,未系统诊治。6年前再次体检肝功能仍异常,ALT 428 U/L,给予保肝对症治疗,1年后停用保肝药物。未规律复诊。1月前无明显诱因出现腹胀,呈进行性加重,当地医院超声提示:肝脏回声增粗,胆囊壁增厚(水肿),脾厚,脾静脉增宽,腹腔积液。为进一步诊治收入我院。发病以来,神志清楚,精神可,食量无变化,睡眠如常,二便正常,体重无明显变化。
流行病史:否认肝病家族史及肝炎接触史,否认乙肝疫苗接种史。否认输血及血制品史,否认其他传染病接触史及疫区居住史。
生长发育正常,目前读小学六年级,成绩优秀。否认长期或特殊药物服用史,否认过敏史。
入院后体格检查:体温:36.3℃,心率:85次/分,呼吸:21次/分,血压:95/60mmHg。神志清楚,精神可,面色可,肝掌可疑,蜘蛛痣阴性。心肺查体未见异常。腹部饱满,软,全腹无压痛、反跳痛,肝脏肋下、剑突下未触及,脾脏肋下2cm,质中,无触痛。肝浊音界大致正常,肝区无叩痛。移动性浊音阳性。双下肢不肿。神经系统未见异常。
入院初步诊断:
肝功能异常
肝硬化可能性大 腹水
病毒性肝炎待除外
遗传代谢性肝病待除外
入院后完善检查:血常规:WBC:4.26E+9/L,HB:115g/L,MCV:89.1fL,%RETIC 2.64%,PLT:97E+9/L;肝功能:ALT:41U/L,AST:66U/L,GGT:111U/L,ALP:170U/L,TBIL:13.9μmol/L,TBA:33.3μmol/L,ALB:25.2g/L,CHE:2183U/L;凝血功能:PTA:37%;病毒学标志物:HAV、HBV、HCV、HEV、EBV、CMV均阴性。自身免疫指标:阴性。免疫球蛋白均正常。铜蓝蛋白:0.037g/L。
腹部CT(见图1):肝硬化伴多发再生结节形成,脾大,腹水,侧支循环形成。
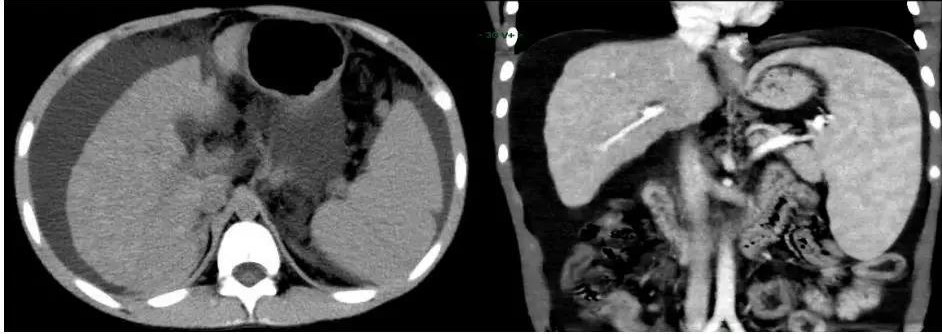
图1.腹部增强CT
眼科检查:K-F环阳性(见图2)。
24小时尿铜定量:1125 μg/24h (正常参考值范围:15-30)
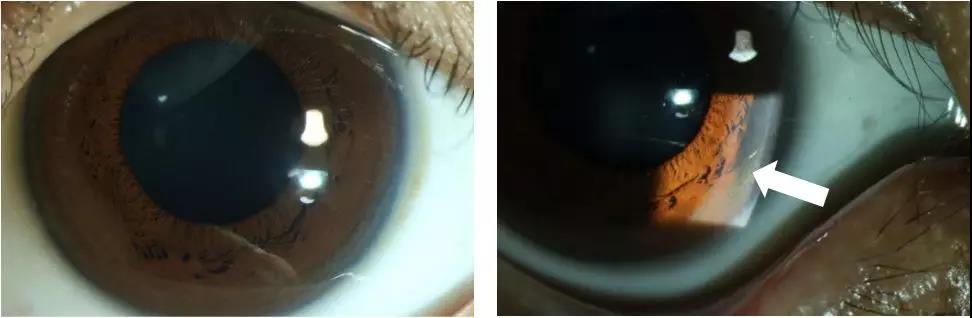
图2.眼睛K-F环阳性
白色箭头所指处:
铜在角膜后弹力层沉积,呈暗棕色
表1.患儿及父母ATP7B基因测序(节选、修订自首都医科大学科技园肝病转化医学研究所检测报告)

根据2001年德国莱比锡Leipzig第八次Wilson病国际会议制定的评分系统:患者K-F环阳性,+2;血清铜蓝蛋白显著降低0.037g/L(低于0.1g/L),+2;24小时尿铜大于2倍ULN,+2;基因检测发现可疑致病突变,+1。总分7分。结合临床,可排除其他肝功异常原因,考虑肝豆状核变性诊断成立。
确定诊断:
肝豆状核变性
慢性肝衰竭
肝硬化失代偿期,合并腹水,侧支循环形成,低蛋白血症
治疗及转归:嘱休息,给予低铜饮食、营养支持指导,以及保肝、补蛋白、利尿等综合对症治疗,并给予青霉胺驱铜治疗,2周后患者肝功能好转,腹水明显消退。目前正在进一步随访中。
郑素军教授:任何不明原因的肝功能异常、肝硬化,都要想到肝豆状核变性的可能,尤其是40岁以下患者,更应警惕。血铜蓝蛋白检测、K-F环检测在多数基层医院已开展,及时检查有利于初筛。有条件者,可以做24小时尿铜测定。肝脏病理改变没有特异性,单独肝脏组织学不能用来确诊Wilson病。只有在临床症状和无创检查不能确诊或怀疑其他或叠加有其他肝脏病变时,才需要肝活检。关于肝组织标本铜染色:由于疾病早期铜主要分布在胞浆中,与金属硫蛋白结合,呈高度水溶性,组织化学方法不能检出,只有到疾病后期铜沉积于肝细胞溶酶体时,罗丹宁或地衣红染色才能检测到溶酶体铜沉积,故不到10%患者可检出灶性铜沉积,铜染色阴性不能排除Wilson病。儿童染色阴性很常见。ATP7B基因检测给肝豆状核变性的诊断提供了强大支持,也有利于对家庭成员进行筛选。
总之,目前肝豆状核变性诊断尚缺乏金标准,较为公认的诊断标准为2001年德国莱比锡Leipzig第八次Wilson病国际会议制定的评分系统,大于等于4分即可诊断。
3 病例供稿及点评专家简介
★ 供稿专家 ★

孔明
博士,副主任医师
首都医科大学附属北京佑安医院
疑难肝病及人工肝中心
佑安专科联盟
遗传代谢性肝病专业委员会秘书
邮箱: kbstef@163.com
★ 点评专家 ★
郑素军 教授
2.1 幻灯(视频)标题及链接:
标题:Wilson病:临床、病理和分子诊断
链接:

长按识别二维码查看幻灯
2.2 幻灯提供专家:郑素军教授
四、专业委员会名单
(按姓氏拼音字母排序)
白洁、白丽、包双宝、边巴央珍、卞丹丹、曾惜秋、常乐、陈芳、陈连清、陈琳、陈悦、程孟怀、程全红、次仁、代东旺、邓雪梅、丁建强、丁向春、杜方雄、段宏宪、段雪飞、段钟平、冯铁柱、高珍、葛迎春、巩维进、苟卫、顾伟玲、郭宁、郭文征、郭小青、何云、胡善雷、黄明星、黄祖雄、霍丽亚、江守伟、焦洪波、经继生、鞠莹、孔明、李宝生、李博、李灿、李广明、李红玲、李建国、李娟、李军、李俊峰、李磊、李秋莲、李荣宽、李响、刘彩峰、刘菲菲、刘晖、刘梅、刘霜、刘冉、刘晓彦、刘耀敏、刘振中、罗磊、吕帅、马玉秀、马臻、苗艳艳、牟丹蕾、南月敏、宁寒冰、牛卫理、彭鹏、乔晓红、邵鸣、盛云建、宋正已、苏丽、孙美艳、覃丽华、谭林、王德步、王飞、王健、王建设、王丽华、王小凤、王艳巧、王仲培、吴刚、吴万锋、吴晓枫、向光明、肖玉珍、谢秋里、易永芬、杨艳玲、于德顺、袁喜先、张定琳、张帆、张立婷、张丽、张丽娟、张缭云、张妍、张银华、张玉山、张志刚、张宗超、赵守松、赵素贤、郑素军、周晓丽、周晓玲、邹桂舟
投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
联系电话:010-63291007
联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间