
本文转载自“佑安肝病感染病专科医疗联盟”
主管:佑安肝病感染病专科医疗联盟(YouMe AID)
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会
指导:段钟平
总编辑:郑素军
本期责任主编:李荣宽
执行编辑:白洁,孔明
本期目录:
一、主编致辞
二、学术进展
三、临床资讯
四、专业委员会名单
五、联系方式
一、主编致辞
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
遗传代谢性肝病月报主要包括以下栏目,力求“一眼抓住最新进展,短时积累临床心得!”
“学术进展”:通过文献分享、专家点评,帮您快速了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座、学会动态等信息,助您感悟遗传代谢性肝病临床诊治之奥妙!
每月一报,让遗传代谢性肝病从罕见病、疑难病变为医者眼中的熟悉病、简单病!
每月一报,助力全面提高我国遗传代谢性肝病的诊治与科研水平!
当前,新型冠状病毒肺炎疫情仍在世界各地肆虐,经过全体医护人员的忘我奋战,我国的疫情控制已经取得了重要胜利,全国肝病医生在这次抗击疫情工作中发挥了重要作用,在这里向全体白衣天使致以最崇高敬意!在段钟平教授和郑素军教授的指导下,感谢孔明教授和白洁博士的辛勤付出,我们遗传代谢性肝病月报第三期准时向大家报到。本期内容在系统回顾关于高胆红素血症的最新文献基础上,为大家分享了关于Dubin-Johnson综合征的2篇最新文献和1例临床病例,希望能够给大家的临床工作带来些许启迪和帮助,在高胆红素血症的诊治工作中多一些思考和参考。
最后,祝愿大家平安健康,祝愿全世界人民抗击疫情早日取得胜利!

李荣宽:男,主任医师,博士研究生导师
大连医科大学附属第二医院感染病科主任
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
二、学术进展
1.Pubmed收录论文汇总归类:2020年2月发表遗传代谢性肝病相关论文共51篇,按遗传代谢性肝病常见分类(比如胆红素、胆汁酸、金属、蛋白、糖等代谢异常),统计结果如下图。
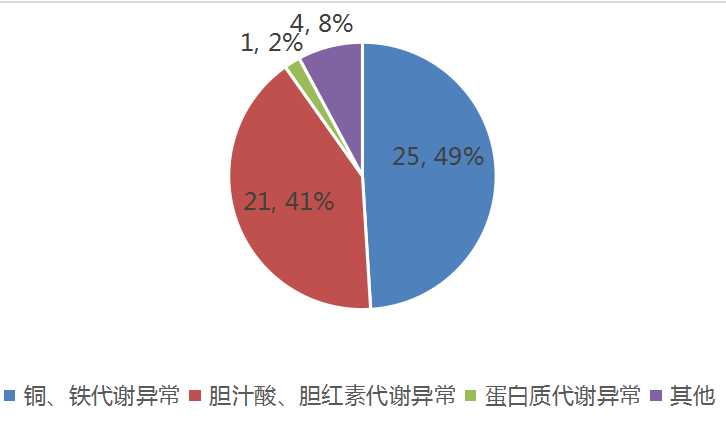
图1.2020年2月Pubmed收录遗传代谢性肝病论文及分类情况
2. PubMed重要文献速览(长按文末二维码或“阅读原文”可下载)
•综述:威尔逊氏病的最新流行情况(Hepatology, 2020, IF=14.971, Q1区)
•组织学评分系统可用于评估胆盐输出泵(BSEP)缺乏症(Hepatol Res, 2020, IF=3.440, Q3区)
•人核受体亚家族1 H组成员4(NR1H4)基因突变导致快速进展的肝衰竭(J Pediatr Gastroenterol Nutr, IF=3.015, Q3区)
•口服和静脉注射放射铁可用于评估血色素沉着症的肝脏铁含量(Int J Mol Sci, IF=4.183, Q2区)
•遗传性血色素沉着症增加关节置换术的发生率(Eur J Gastroenterol Hepatol, IF=2.198, Q4区)
•血色素沉着症的基因突变与阿尔茨海默病无关(Neurobiol Aging, IF=4.398, Q2区)
•TJP2基因的纯合突变c.3334C>T可导致进行性家族性肝内胆汁淤积症-4(PFIC-4)PFIC-4(World J Gastroenterol, IF=3.411, Q3区)
•我国由首例HFE基因复合杂合突变p.C282Y/p.R71X导致的血色素沉着症(Front Genet, IF=3.517, Q2区)
•血清间接胆红素浓度和帕金森病及其运动亚型有关(Neurodegener Dis, IF=2.798, Q3区)

•胆红素通过增加酸敏感离子通道活性加重小鼠模型神经系统损伤(Sci Transl Med, IF=17.161, Q1区)
3.Pubmed精选文献介绍及专家简评(长按文末二维码或“阅读原文”可下载文献全文)
3.1文献一(英文): Corpechot C, Barbu V, Chazouillères O,et al. Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited
cholestatic disorders. Liver Int, 2020;40(1):163-174. Impact factor= 5.542, Q2区。
Corpechot C, Barbu V, Chazouillères O,et al. Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited
cholestatic disorders. Liver Int, 2020;40(1):163-174. Impact factor= 5.542, Q2区。
文献一(中文):Corpechot C, Barbu V, Chazouillères O等。ABCC2对Dubin-Johnson综合征和遗传性胆汁淤积性疾病的遗传贡献.国际肝病, 2020; 40(1):163-174. 影响因子= 5.542, Q2区。
英文摘要:
BACKGROUND AND AIMS:
The ABCC2 gene is implicated in Dubin-Johnson syndrome (DJS), a rare autosomal recessive liver disorder. The primary aim of this study was to determine the diagnostic value of ABCC2 genetic testing in the largest cohort of DJS reported to date. The high number of patients with cholestatic manifestations in this series prompted us to evaluate the genetic contribution of rare, potentially pathogenic ABCC2 variants to other inherited cholestatic disorders.
METHODS:
The cohort study included 32 patients with clinical DJS diagnosis, and 372 patients referred for the following disorders: low phospholipid-associated cholelithiasis (LPAC) syndrome, intrahepatic cholestasis of pregnancy (ICP) and benign recurrent intrahepatic cholestasis (BRIC). ABCC2 was screened by next-generation sequencing.
RESULTS:
Most patients with clinical DJS had positive genetic diagnosis (n = 30; 94%), with a great diversity of point mutations and copy number variations in ABCC2. Strikingly, eight (27%) of these patients showed transient cholestatic features at presentation: four neonatal cholestasis, two ICP, one contraceptive-induced cholestasis and one sporadic cholestasis. Conversely, the frequency of rare, heterozygous, potentially pathogenic ABCC2 variants in patients with LPAC, ICP or BRIC did not differ significantly from that of the general population.
CONCLUSIONS:
This large series reveals that DJS is a highly homogeneous Mendelian disorder involving a large spectrum of ABCC2 variants. Genetic testing is crucial to establish early DJS diagnosis in patients with atypical presentations, such as neonatal cholestasis. This study also provides no evidence for the contribution of rare, potentially pathogenic ABCC2 variants to other inherited cholestatic disorders.
中文摘要:
背景和目的:
ABCC2基因与Dubin-Johnson综合征(DJS)发生有关,这是一种罕见的常染色体隐性遗传性肝病。这项研究的主要目的是,确定ABCC2基因检测在目前为止的、最大DJS队列中的诊断价值。该队列中有大量胆汁淤积表现的患者,这促使我们评估这种罕见的、潜在致病性的ABCC2变异在其他遗传性胆汁淤积性疾病发病中的作用。
方法:
该队列研究包括32例临床诊断的DJS患者,以及372例因以下疾病而转诊的患者:低磷脂相关性胆石症综合征(LPAC),妊娠肝内胆汁淤积症(ICP)和良性复发性肝内胆汁淤积症(BRIC)。通过二代测序检测ABCC2变异。
结果:
大多数临床诊断DJS的患者遗传学诊断也为阳性(n = 30;94%),但ABCC2基因的点突变和拷贝数变异具有很大的多样性。令人惊讶的是,这些患者中有8名(27%)表现为短暂的胆汁淤积特征:4例新生儿胆汁淤积,2例ICP,1例避孕药致胆汁淤积和1例偶发性胆汁淤积。相反,在LPAC,ICP或BRIC的患者中,罕见的、杂合的、潜在致病的ABCC2变异的频率与一般人群的频率没有显着差异。
结论:
这个大型队列揭示了DJS是一种高度同质的孟德尔疾病,涉及ABCC2基因的多种变异。基因检测对于具有非典型表现(例如新生儿胆汁淤积)的DJS的早期诊断至关重要。这项研究也表明罕见的、潜在致病性ABCC2变异对其他遗传性胆汁淤积性疾病没有贡献。
李荣宽教授简评:
Dubin-Johnson 综合征( DJS) 是一种少见的以慢性持续性或间歇性黄疸为临床特点的常染色体隐性遗传病,常见于青少年和幼年。因为ABCC2 /MRP2(10q24)基因缺陷,使肝细胞中结合胆红素及其他有机阴离子向毛细胆管排泄障碍,引起血清结合胆红素升高。肝脏表现为“黑肝”,组织学可见肝小叶中央区肝细胞溶酶体内棕褐色色素颗粒沉着。
为进一步明确ABCC2基因突变在DJS发病和诊断中的价值,本研究对临床诊断DJS的患者和其它遗传相关的胆汁淤积性疾病患者通过二代测序检测ABCC2变异情况,发现临床诊断DJS的患者遗传学诊断的阳性率高达94%,而其他遗传性胆汁淤积性疾病患者中ABCC2的变异频率与一般人群没有显着差异。这一研究是目前纳入DJS样本量最大的队列研究,其结果证明了基因检测对于诊断DJS至关重要,可以通过检测ABCC2变异来与其他遗传性胆汁淤积性疾病相鉴别。
本研究优点:首先,这项研究纳入的DJS样本量是目前最大的,达到32例,另外提供了372例遗传性胆汁淤积性疾病做对照,对照组包括LPAC、ICP、BRIC,这对于遗传代谢性疾病等少见病来说非常难得,研究结果对临床提供的指导意义很大。其次,传统DSJ的诊断主要靠临床表现及病理检查,即使被奉为“金标准”的病理检查,在DSJ早期病变不典型时,也可能造成诊断困难。本研究应用二代测序方法检测ABCC2基因突变,明确了该基因在DSJ中具有很高的突变率,而其它遗传性胆汁淤积性疾病黄疸患者的该基因突变率与一般人群没有显著差异,不但为揭示DSJ的发病机制提供帮助,更为该病的确诊提供了更精确的指标。
本研究不足:现代遗传学的发展为遗传性胆汁淤积性疾病揭示病因以及发病机制提供了重要作用,本研究纳入了多种遗传相关的胆汁淤积性疾病作对照,如果能够纳入其他以结合胆红素升高为主的疾病则更完善,例如与DJS在临床上曾被认为是一个疾病的Rotor综合征在本研究中没有被纳入。
可供借鉴或有所启迪之处:目前二代测序技术已经在临床工作中得到推广,不明原因黄疸患者进行基因检测有助于疾病的尽早确诊。
3.2 文献二(英文):Li Y, Li Y, Yang YW, et al. Next generation sequencing reveals co-existence of hereditary
spherocytosis and Dubin-Johnson syndrome in a Chinese girl: A case report. World J Clin Cases, 2019 Oct 26;7(20):3303-3309. Impact
factor=1.153, Q4。
文献二(中文):Wooton-Kee CR, Robertson M, Zhou Y等.二代测序揭示了中国女孩的遗传性球形细胞增多症合并Dubin-Johnson综合征:一个病例报道. 世界临床病例杂志,2019 Oct 26;7(20):3303-3309.影响因子=1.153, Q4区。
英文摘要:
BACKGROUND:
Hereditary spherocytosis (HS) is a hereditary disease of hemolytic anemia that occurs due to the erythrocyte membrane defects. Dubin-Johnson syndrome (DJS), which commonly results in jaundice, is a benign hereditary disorder of bilirubin clearance that occurs only rarely. The co-occurrence of HS and DJS is extremely rare. We recently diagnosed and treated a case of co-occurring HS and DJS.
CASE SUMMARY:
A 21-year-old female patient presented to our department because of severe jaundice, severe splenomegaly, and mild anemia since birth. We eventually confirmed the diagnosis of co-occurring DJS and HS by next generation sequencing (NGS). The treatment of ursodeoxycholic acid in combination with phenobarbital successfully increased hemoglobin and reduced total bilirubin and direct bilirubin.
CONCLUSION:
The routine application of NGS can efficiently render a definite diagnosis when inherited disorders are suspected.
中文摘要:
背景:
遗传性球形红细胞增多症(HS)是一种遗传性溶血性贫血,是由红细胞膜缺陷所致。Dubin-Johnson综合症(DJS)通常会导致黄疸,是一种罕见的、良性的遗传性胆红素清除障碍。HS和DJS并存的情况极为罕见。我们最近诊断并治疗了一例HS合并DJS的病例。
案例摘要:
一名21岁的女性患者,自出生后就有严重的黄疸,严重的脾肿大和轻度贫血。我们最终通过二代测序(NGS)证实了并存的DJS和HS。熊去氧胆酸联合苯巴比妥治疗成功升高了血红蛋白,并降低总胆红素和直接胆红素。
结论:
当怀疑遗传性疾病时,NGS的常规应用可以有效地做出明确诊断。
李荣宽教授简评:
Dubin-Johnson 综合征( DJS)患者的肝细胞对胆红素的摄入、转运、与葡萄糖醛酸的结合过程是正常的,但毛细胆管ABCC2 /MRP2基因缺陷,导致结合型胆红素向毛细胆管内分泌的功能降低,引起血清中结合胆红素升高。但如果患者合并一些导致非结合胆红素和/或结合胆红素升高的疾病,例如遗传性球形红细胞增多症、病毒性肝炎、胆道结石等,会对临床诊断带来干扰,造成DJS的漏诊。临床工作中最简单有效的鉴别思维是当患者的黄疸无法用一种疾病来解释,如这篇文章中所报道的遗传性球形红细胞增多症,该种疾病的黄疸性质应该是非结合胆红素升高,临床发现结合胆红素也明显升高时,应当想到是否合并其它疾病。或者在明确诊断的疾病得到有效治疗后,以结合胆红素升高为主要表现的黄疸仍持续存在或反复发生,也应当考虑是否存在DJS等其它疾病,必要时应进行肝脏组织活检,在有条件时进行基因测序,明确突变基因对于DJS的诊断非常关键。
4.本期Pubmed文献编译及短评专家简介
4.1 文献编译

白洁:首都医科大学附属北京佑安医院在读博士研究生
佑安专科联盟遗传代谢性肝病专业委员会秘书
邮箱:docbai@yeah.net
4.2 短评专家
李荣宽:男,主任医师,博士研究生导师
大连医科大学附属第二医院感染病科主任
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
三、临床资讯
1.典型病例分享
1.1 病例简介(Dubin-Johnson综合征):
患者高某某,女,51岁,主因“尿黄10年余,肝功异常2年。”门诊以“肝功异常”于2015年4月27日收住院。
现病史:患者10年余前无明显诱因出现浓茶色尿。未予重视,后逐渐加重。2年前体检发现肝功异常,谷丙转氨酶正常,总胆红素升高82umo/L,直接胆红素升高,具体不详,未予特殊治疗。10月前给予水飞蓟、茴三硫等治疗,复查肝功基本正常,停药近1月,此后间断复查总胆红素升高,再次服用水飞蓟、茴三硫治疗。5月前复查总胆红素109.7 umol/L,服用熊去氧胆酸胶囊治疗,服药2月余,3月前出现眼睑、眉部瘙痒、伴红色皮疹,停用上述药物治疗。2月前于当地医院住院,化验总胆红素60.2 umol/L,给子保肝治疗,患者总胆红素降至49.2umo/L出院,2周前复查总胆红素反弹至56.6umol/L,自觉厌油腻,乏力,进食量略减少,尿黄如浓茶,无皮肤瘙痒,无陶土样大便。为进步诊治收住院。
既往史:22年前因胆囊结石行胆囊切除术。有长期间断染发情况。家族史:家族中无肝病或类似病史者。
入院查体:体温:36.3℃,心率85次/分,血压:110/70mmHg,呼吸:17次/次。神志清楚,皮肤巩膜中度黄染,肺呼吸音正常,心律齐,腹软,无压痛、反跳痛及肌紧张,肝脏、脾脏未触及,肝区无叩痛,移动性浊音阴性,下肢水肿无下肢水肿,神经系统未见异常。
入院诊断:黄疸原因待查
入院后检查结果:血常规:WBC:4.97E+9/L,N:54.6%,HGB: 157g/L,PLT:293 E+9/L。尿常规:尿胆红素(+),余正常。肝功能:ALT:65U/L,AST30U/L,TBIL:48.9umol/L,DBil:34.6 umol/L,ALB:47.5g/L,ALP:110U/L,GGT:48U/L,TBA:8.2umol/L,CHE:13150U/L。血脂:TG:1.72mmol/L,CHOL:5.3mmol/L,LDL-C:3.19mmol/L。肾功能正常。嗜肝病毒相关检查均(-),自身抗体(-),免疫球蛋白+补体:正常。铜蓝蛋白:正常。血清铁、铁蛋白、总铁结合力均正常。腹部超声:弥漫性肝病表现。
为进一步明确诊断,行肝穿刺活检病理检查。
镜下所见:小叶结构存在,腺泡III带灶性肝细胞大泡性脂肪变(约2%),肝细胞内大量色素颗粒沉积,以小叶中心明显,肝实质散在点状坏死,汇管区少量炎细胞浸润,未见明显纤维化。
免疫组化:HBsAg(-),HBcAg(-),CK7(胆管+),CK19(胆管+)
病理诊断:结合临床,考虑Dubin-Johnson综合征可能。
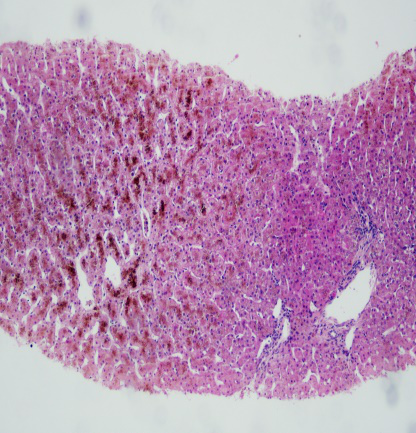
HE 100×
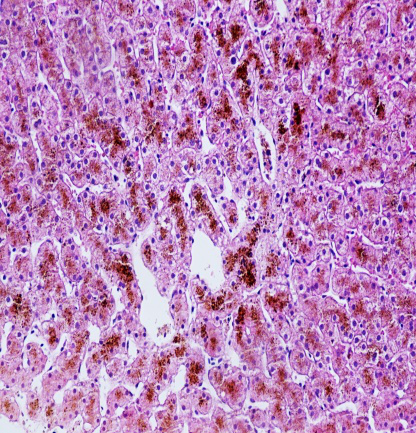
HE 200×
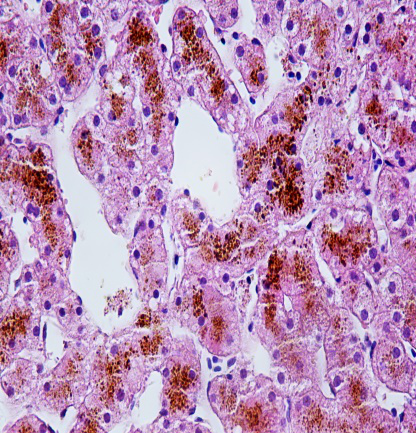
HE 400×
临床诊断:Dubin-Johnson综合征
治疗:给予保肝退黄对症支持等综合治疗,患者肝功能好转。
1.2 专家点评
李荣宽教授:Dubin-Johnson综合征(DJS)是临床上较为罕见的常染色体隐性遗传性疾病,1954年由Dubin和Johnson首先报道。目前,其发病机制认为是位于10q24的基因( ABCC2 /MRP2) 缺陷,导致毛细胆管的多药耐药相关蛋白异常,使肝细胞中结合胆红素及其他有机阴离子向毛细胆管排泄障碍。DJS临床诊断依据主要包括: (1)青少年发病,常有家族史;(2)长期持续或间歇性轻中度黄疸,患者一般情况良好;(3)以结合胆红素升高为主,其它肝功能指标基本正常;(4)口服胆囊造影不显影或显影淡;(5)肝组织活检:肉眼观肝组织外观呈绿黑色,镜下见肝细胞胞浆内有较多大小不一的棕褐色色素颗粒。另外,尿粪卟啉异构体测定、磺溴酞钠试验、核素肝胆显像等检查对DJS的诊断也有一定帮助。
本例患者中年女性,黄疸病史长达10余年,表现为反复发作性黄疸,以直接胆红素增高为主,不伴有其它症状与体征,就诊于多家医院,一直未能明确诊断。此次住院期间完善了与胆红素升高相关的多项检查:嗜肝病毒相关检查排查病毒性肝炎;自身抗体、免疫球蛋白排查自身免疫性肝病;铜蓝蛋白排查Wilson病;血清铁、铁蛋白、总铁结合力排查血色病等等,结果均阴性,病因仍扑朔迷离。为明确诊断,进行了肝组织活检,镜下见:肝细胞内大量色素颗粒沉积,提示DJS。至此,结合该患者如此长的病史,只以直接胆红素升高为临床表现,不伴有肝脏和其他脏器明显损伤,DJS临床诊断应该成立。但也存在缺憾,没能为患者和其家人进行基因检测。因为基因测序技术的进步以及人们对遗传代谢性肝病认识的加深,突变基因的检测在这类疾病的诊断中所起的决定性价值越来越受到临床的重视。
另外,这个病例还有一些其它应当引起我们注意的问题:首先患者22年前因胆囊结石行胆囊切除术,这影响我们进行口服胆囊造影检查,同时我们应当想到是否存在肝内外胆管结石造成胆汁排泌不畅而引起胆汁淤积性黄疸,当然,这个问题已经被腹部超声检查除外了。还有,这个患者有长期间断染发史,腹部超声提示“弥漫性肝病表现”,肝组织学见到“腺泡III带灶性肝细胞大泡性脂肪变(约2%),肝实质散在点状坏死”。这些情况提醒我们这个患者是否存在药物相关性肝损伤(DILI)的可能。
1.3 病例供稿及点评专家简介
1.3.1供稿专家:

韩莹:女,医学博士,主任医师
首都医科大学附属北京佑安医院肝病免疫科
1.3.2 点评专家:李荣宽教授
2.讲座或幻灯分享
2.1幻灯标题及连接:先天性黄疸的分子诊断
2.2幻灯提供专家:郑素军教授

郑素军:博士,主任医师
首都医科大学附属北京佑安医院疑难肝病及人工肝中心
佑安专科联盟遗传代谢性肝病专业委员会主任委员
四、专业委员会名单(按拼音排序):
白洁、白丽、包双宝、边巴央珍、卞丹丹、曾惜秋、常乐、陈芳、陈连清、陈琳、陈悦、程孟怀、程全红、次仁、代东旺、邓雪梅、丁建强、丁向春、杜方雄、段宏宪、段雪飞、段钟平、冯铁柱、高珍、葛迎春、巩维进、苟卫、顾伟玲、郭宁、郭文征、郭小青、何云、胡善雷、黄明星、黄祖雄、霍丽亚、江守伟、焦洪波、经继生、鞠莹、孔明、李宝生、李博、李灿、李广明、李红玲、李建国、李娟、李军、李俊峰、李磊、李秋莲、李荣宽、李响、刘彩峰、刘菲菲、刘晖、刘梅、刘霜、刘冉、刘晓彦、刘耀敏、刘振中、罗磊、吕帅、马玉秀、马臻、苗艳艳、牟丹蕾、南月敏、宁寒冰、牛卫理、彭鹏、乔晓红、邵鸣、盛云建、宋正已、苏丽、孙美艳、覃丽华、谭林、王德步、王飞、王健、王建设、王丽华、王小凤、王艳巧、王仲培、吴刚、吴万锋、吴晓枫、向光明、肖玉珍、谢秋里、易永芬、杨艳玲、于德顺、袁喜先、张定琳、张帆、张立婷、张丽、张丽娟、张缭云、张妍、张银华、张玉山、张月荣、张志刚、张宗超、赵守松、赵素贤、郑素军、周晓丽、周晓玲、邹桂舟
五、联系方式
投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
联系电话:010-63291007
联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间