
本文转载自“佑安肝病感染病专科医疗联盟”
本期目录:
一、主编致辞
二、学术进展
三、临床资讯
四、专业委员会名单
五、联系方式
一、主编致辞
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
遗传代谢性肝病月报主要包括以下栏目,力求“一眼抓住最新进展,短时积累临床心得!”
“学术进展”:通过文献分享、专家点评,帮您快速了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座、学会动态等信息,助您感悟遗传代谢性肝病临床诊治之奥妙!
每月一报,让遗传代谢性肝病从罕见病、疑难病变为医者眼中的熟悉病、简单病!
每月一报,助力全面提高我国遗传代谢性肝病的诊治与科研水平!
遗传代谢性肝病是基因缺陷而导致代谢异常的一大类疾病,有家族聚集特点,虽然单病种发病率很低,但因种类繁多,故总体发病率较高,所以临床上各种遗传代谢性肝病仍不容忽视。尤其是,有些疾病早期发现及时干预是能够避免严重的不良结局的。在段钟平教授和郑素军教授的指导下,遗传代谢性肝病月报每月一期,尤其是在新冠疫情肆虐的情况下,仍坚持如期完成,在此要感谢孔明教授和白洁博士的辛勤付出!本期月报主要内容为遗传性肝内胆汁淤积,从最新研究进展到典型临床病例分享,为后续诊治此类疾病提供了更多的方法和思路。
在此,祝愿大家平安健康,工作顺利!祝愿全世界疫情早日得到控制!还一个健康的生活空间给人类!

张缭云:女,主任医师,教授,硕士研究生导师
山西医科大学第一医院感染科主任
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
二、学术进展
1.Pubmed收录论文汇总归类:2020年3月发表遗传代谢性肝病相关论文共29篇,按遗传代谢性肝病常见分类(比如胆红素、胆汁酸、金属、蛋白、糖等代谢异常),统计结果如下图。
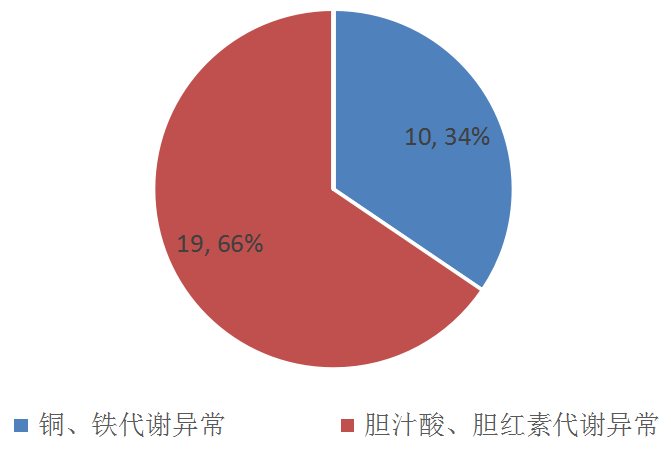
图1.2020年3月Pubmed收录遗传代谢性肝病论文及分类情况
2. PubMed重要文献速览(长按文末二维码或“阅读原文”可下载)
□血浆胆汁酸谱可监测部分内部胆汁分流(PIBD )治疗PFIC2患者的有效性(Ann Transl Med, 2020, IF=3.689, Q2区)

□进行性家族性肝内胆汁淤积症可导致胆固醇升高(J Pediatr Endocrinol Meta, 2020, IF=1.239, Q4区)
□遗传性血色素沉着症可导致特发性脑钙化(BMC Neurol, 2020, IF=1.239, Q3区)
□综述:铁代谢和肿瘤进展(Int J Mol Sci, 2020, IF=4.183, Q2区)
□利用小基因可预测UGT1A1基因剪切突变的功能(Front Genet, 2020, IF=3.517, Q2区)
□ABCC2突变p.G693R导致中国人群Dubin-Johnson综合征(Orphanet J Rare Dis, 2020, IF=3.687, Q2区)
□弥散张量成像发现I型Crigler-Najjar综合征的深部灰质和白质区域的微结构变化(J Comput Assist Tomogr, 2020, IF=1.301, Q4区)
□发现I型Crigler-Najjar综合征新碱基对重复突变c.55_76dup(BMC Gastroenterol, 2020, IF=2.252, Q3区)
□成人肝豆状核变性急性肝损伤的预后:不接受肝移植的存活率高(Liver Transpl, 2020, IF=4.159, Q1区)
□辅助抗氧化治疗改善神经系统威尔逊病的临床结局(J Mol Neurosci, 2020, IF=2.577, Q3区)
3.Pubmed精选文献介绍及专家简评(点击“下载全文”可下载文献全文)
3.1文献一(英文):Weber ND, Odriozola L, Martínez-García J, et al. Gene therapy for progressive familial intrahepatic cholestasis type 3 in a clinically relevant mouse model. Nat Commun, 2019 13;10(1):5694. Impact factor=11.878, Q1区。
文献一(中文):Weber ND, Odriozola L, Martínez-García J等。基因治疗进行性家族性肝内胆汁淤积3型小鼠模型. 自然通讯, 2019 13;10(1):5694. 影响因子=11.878, Q1区。
英文摘要:
Progressive familial intrahepatic cholestasis type 3 (PFIC3) is a rare monogenic disease caused by mutations in the ABCB4 gene, resulting in a reduction in biliary phosphatidylcholine. Reduced biliary phosphatidylcholine cannot counteract the detergent effects of bile salts, leading to cholestasis, cholangitis, cirrhosis and ultimately liver failure. Here, we report results from treating two- or five-week-old Abcb4-/- mice with an AAV vector expressing human ABCB4, resulting in significant decreases of PFIC3 disease biomarkers. All male mice achieved a sustained therapeutic effect up through 12 weeks, but the effect was achieved in only 50% of females. However, two-week-old females receiving a second inoculation three weeks later maintained the therapeutic effect. Upon sacrifice, markers of PFIC3 disease such as, hepatosplenomegaly, biliary phosphatidylcholine and liver histology were significantly improved. Thus, AAV-mediated gene therapy successfully prevented PFIC3 symptoms in a clinically relevant mouse model, representing a step forward in improving potential therapy options for PFIC3 patients.
中文摘要:
进行性家族性肝内胆汁淤积症3型(PFIC3)是一种罕见的单基因疾病,由ABCB4基因突变引起,导致胆汁磷脂酰胆碱减少。胆汁磷脂酰胆碱的减少不能抵消胆盐的去垢作用,从而导致胆汁淤积、胆管炎、肝硬化,并最终导致肝衰竭。本文中,我们使用人ABCB4基因的AAV载体治疗两周或五周大的Abcb4-/-小鼠,治疗后PFIC3疾病的生物标记物显著降低。所有雄性小鼠在12周内都达到了持续的治疗效果,但只有50%的雌性达到了这种效果。三周后接受第二次接种的两周大的雌性也有持续的治疗效果。处死小鼠后,发现PFIC3疾病的标志物,例如肝脾肿大、胆汁磷脂酰胆碱和肝脏组织学得到了明显改善。因此,AAV介导的基因治疗成功地预防了PFIC3小鼠模型的疾病相关症状,是未来治疗PFIC3的潜在选择。
张缭云教授简评:
PFIC3是由一种ABCB4基因突变导致的遗传性胆汁淤积性肝病,常在婴幼儿发病。ABCB4基因编码蛋白MDR3,负责将磷脂从肝细胞转运到胆小管中。MDR3功能降低或缺乏时,胆汁中磷脂减少或缺乏,不能抵消胆盐的去污作用,游离胆盐损伤胆管细胞,出现胆汁淤积。
本研究通过腺病毒介导的ABCB4基因治疗PFIC3小鼠模型,雄性小鼠在12周内达到了持续的治疗效果,而雌性小鼠接受第二次接种后也能到达持续的治疗效果。治疗后多种生物学标志物均显著改善,包括血清肝转氨酶、碱性磷酸酶、胆汁酸和胆红素等血清学指标,肝脾大,胆汁中磷脂酰胆碱浓度,以及肝脏组织学异常,例如纤维化、胶原蛋白沉积和细胞浸润。而且组织学检查发现治疗后肝纤维化程度显著降低至几乎完全消失,这是治疗有效最明确的证据。
尽管目前基因治疗可能涉及到细胞恶变、免疫反应等安全性问题,和体内表达基因的可控性问题,甚至在伦理上有很大争议。但基因治疗仍然是家族性进行性肝内胆汁淤积等严重遗传代谢性疾病的潜在治疗方法,有望彻底治愈患者,提高他们的生活质量。
3.2 文献二(英文):Zhang J, Yang Y, Gong JY, et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: clinical, histological, and ultrastructural characterization. Liver Int. 2020 Mar 2. Impact factor=5.542, Q2区。
文献二(中文):Zhang J, Yang Y, Gong JY等. 和USP53双等位基因变异有关的低GGT肝内胆汁淤积:临床、组织学和超微结构特点. 国际肝病,2020 Mar 2.影响因子=5.542, Q2区。
英文摘要:
BACKGROUND AND AIMS:
In about 20% of children with cholestasis and normal or low serum gamma-glutamyltransferase (GGT) activity, no etiology is identified. We sought new genes implicated in pediatric hepatobiliary disease.
METHODS:
We conducted whole exome sequencing in 69 children evaluated at our center from 2011 to 2018 who had low-GGT cholestasis and in whom homozygous / compound heterozygous predictedly pathogenic variants (PPV) in ATP8B1, ABCB11, NR1H4, MYO5B, or TJP2 were not found. Clinical records and findings on light microscopy and transmission electron microscopy of liver-biopsy materials were reviewed.
RESULTS:
In 7 patients from 7 unrelated families, biallelic PPV (10 in total) were found in USP53, recently associated with intrahepatic cholestasis. Seven variants were classified as pathogenic: one canonical splicing, c.569+2T>C, and six nonsense or frameshifting: c.169C>T (p.Arg57Ter), c.581delA (p.Arg195GlufsTer38), c.831_832insAG (p.Val279GlufsTer16), c.1012C>T (p.Arg338Ter), c.1426C>T (p.Arg476Ter), and c.1558C>T (p.Arg520Ter). Three were likely pathogenic: c.297G>T (p.Arg99Ser), c.395A>G (p.His132Arg), and c.878G>T (p.Gly293Val). In all patients, jaundice began at age <7mo. Cholestasis was transient, with documented resolution of hyperbilirubinemia in all (oldest patient now aged 5y) except one, who was lost to follow-up. Light microscopy identified intralobular cholestasis, giant-cell change of hepatocytes, and perisinusoidal-perihepatocytic and portal-tract fibrosis. Ultrastructural study revealed elongated hepatocyte-hepatocyte tight junctions. One patient was deaf.
CONCLUSION:
USP53 interacts with the tight-junction constituent TJP2. TJP2 mutation can cause low-GGT intrahepatic cholestasis, with elongated hepatocyte-hepatocyte tight junctions, as well as deafness. Our findings extend a preliminary report of USP53 disease and indicate that USP53 mutation may generate a partial phenocopy of TJP2 disease.
中文摘要:
背景和目的:
约20%的正常或较低水平血清γ-谷氨酰转移酶(GGT)的胆汁淤积儿童,尚没有找到病因。我们希望寻找与小儿肝胆疾病有关的新基因。
方法:
我们对2011年至2018年在我们中心评估的69名儿童进行了全外显子组测序。这些儿童均患有低GGT胆汁淤积症,并且未检测到纯合或复合杂合的ATP8B1、ABCB11、NR1H4、MYO5B或TJP2致病突变。本文回顾了这些患儿的临床资料和肝活检的光镜、电镜结果。
结果:
在来自7个无关家庭的7例患者中,发现了USP53的双等位基因致病突变(共10个突变),该基因和肝内胆汁淤积有关。其中七个变异都是致病性的:一个是标准剪接c.569 + 2T> C,六个是无义突变或移码突变:c.169C> T(p.Arg57Ter)、c.581delA(p.Arg195GlufsTer38)、c.831_832insAG(p Val279GlufsTer16)、c.1012C> T(p.Arg338Ter)、c.1426C> T(p.Arg476Ter)和c.1558C> T(p.Arg520Ter)。还有三个变异是可能致病:c.297G> T(p.Arg99Ser)、c.395A> G(p.His132Arg)和c.878G> T(p.Gly293Val)。所有患者黄疸发病时间小于7个月,而且胆汁淤积都是一过性的。除一名失访者外,所有患者(年龄最大的患者5岁)高胆红素血症均可缓解。光镜下可见小叶内胆汁淤积、肝细胞巨细胞变化、窦周肝细胞和门脉纤维化。超微结构显示,肝细胞之间的紧密连接延长。有一名患者失聪。
结论:
USP53可与紧密连接的成分TJP2相互作用。 TJP2突变导致低GGT肝内胆汁淤积,伴肝细胞之间的紧密连接延长以及耳聋。我们的发现初步表明了USP53疾病的内容,并提示USP53突变可能产生TJP2疾病的部分表型。
张缭云教授简评:
USP53编码泛素羧基末端水解酶53(USP53),属于去泛素化酶家族。既往在小鼠耳聋模型中已经证明,同源蛋白Usp53可与紧密连接的紧密连接蛋白2(Tjp2,由Tjp2编码)相互作用。而Tjp2蛋白参与听毛细胞和听力的存活,调节细胞间紧密连接。Tjp2双等位基因突变可以导致耳聋、低GGT的家族性进行性肝内胆汁淤积4型(PFIC4)。而本研究中的儿童既有低GGT肝内胆汁淤积的表现,而且有一名儿童出现耳聋,与既往研究结果一致,再次证明USP53和Tjp2这两个基因之间可能具有相互作用。也提示了USP53基因突变会导致和Tjp2疾病类似的表型。
本研究优点:1.样本量较大,入排标准明确。本研究共纳入了69名正常或较低水平血清γ-谷氨酰转移酶的胆汁淤积儿童,均进行全外显子组测序。所有患者均除外了感染、药物性肝损伤、自身免疫性肝炎和胆道闭锁。纳入标准为:1)如果总胆红素(TB)>5mg/dL,直接胆红素(DB)大于总胆红素的20%;如果TB<5mg/dL,DB>1mg/dL,并且总胆汁酸显着升高,没有明确的肝胆疾病;2)血清GGT值低(最高GGT<100IU/L);3)ATP8B1、ABCB11、NR1H4、TJP2和MYO5B基因均未发现致病突变。将接受基因测序的其他肝脏疾病(原因不明的高GTT胆汁淤积症、转氨酶升高或肝硬化)和非肝脏疾病的患者当作对照。2.综合判断突变的致病性。在这些儿童中发现了共10个USP53基因突变。根据等位基因频率在千人基因组和ExAC项目中低于0.01;无义突变、移码突变、剪切突变或基因预测软件预测为致病突变;当为双等位基因突变时,仅存在于低GGT胆汁淤积患者这三项要求,判断基因突变的致病性。最终确定了7个和低GGT肝内胆汁淤积有关的USP53致病突变。
本研究的不足之处:缺乏对基因突变进行功能验证,如能在细胞水平进行酶活性检测、动物模型上验证等,则更有说服力。
可供借鉴或有所启迪之处:目前二代测序技术已经在临床工作中得到推广,积极进行基因检测,有助于疾病诊断和发现新的致病基因。
4.本期Pubmed文献编译及短评专家简介
4.1 文献编译

白洁:首都医科大学附属北京佑安医院在读博士研究生
佑安专科联盟遗传代谢性肝病专业委员会秘书
邮箱:docbai@yeah.net
4.2 短评专家
张缭云:女,主任医师,教授,硕士研究生导师
山西医科大学第一医院感染科主任
佑安专科联盟遗传代谢性肝病专业委员会副主任委员
三、临床资讯
1.典型病例分享
1.1 病例简介(BRIC1型):
患者,男性,23岁,主因“间断肝功能异常3年余”门诊以“黄疸”于2019年3月19日收住院。
患者3年余前无明显诱因出现皮肤、巩膜轻度黄染,伴皮肤瘙痒、尿色加深,无灰白便。偶有恶心,无呕吐。无发热、腹痛、黑便、神志改变等症状。就诊于河南省济源市人民医院,发现肝功能异常,具体不详,给予保肝、降酶及退黄治疗后,病情无好转,总胆红素升至约320μmol/L。后进一步就诊于河南省人民医院,自诉总胆红素最高可达600μmol/L,考虑“胆汁淤积性肝炎”,继续予保肝、降酶及退黄等治疗,并予行人工肝治疗3次,治疗后总胆红素下降至正常后出院。出院后未再规律复查。半年余前,患者再次出现胆红素升高,最高升至约380μmol/L,就诊于新乡医学院,再次行人工肝治疗2次后好转。出院后口服保肝药物治疗,2月余前,总胆红素降至正常。1月余前,患者再次发现皮肤、巩膜黄染,复查肝功能示总胆红素162.7μmol/L,现为明确病因,收入我科。
患者自发病以来精神可、食量无变化、睡眠无改变、小便色深,呈茶色、大便正常,体重1个月减轻3kg。
既往:有血浆使用史。否认过敏史。
家族史:家族中无肝病或类似病史者。
个人史:吸烟6年,日均吸烟3-4支,戒烟6个月,偶尔饮酒。
入院查体:体温:36.4℃,心率84次/分,血压:118/88mmHg,呼吸:20次/分。神志清楚,精神状态可,皮肤巩膜中度黄染,肝掌阳性,未见蜘蛛痣及毛细血管扩张。双肺呼吸音正常。心律齐,未及病理性杂音。腹软,无压痛、反跳痛及肌紧张,肝脏、脾脏未触及,肝区无叩痛,移动性浊音阴性,双下肢无水肿,神经系统未见异常。
入院诊断:肝功能异常原因待查 遗传性肝病可能性大 自身免疫性肝病待除外 低白蛋白血症
入院后检查结果:血常规:WBC:8.66E+9/L,N:71.6%,HGB: 102g/L,PLT:464E+9/L。尿常规:RBC 偶见,余正常。肝功能:ALT 42U/L,AST 28U/L,TBIL 199.4μmol/L,DBil 151.3μmol/L,ALB 43.9g/L,ALP 159U/L,GGT 25U/L,TBA 163.5μmol/L,CHE 4422U/L。NH3 66ug/dL。凝血功能:正常 PT 11.7s PTA 93% INR 1.04 APTT 33S。血脂、肾功能正常。嗜肝病毒相关检查均(-),自身抗体(-),抗线粒体抗体M2亚型抗体(-),肝抗原谱(-),免疫球蛋白+补体:正常。铜蓝蛋白:正常。24小时尿酮:104.9ug/24小时。血清铁、铁蛋白、总铁结合力均正常。腹部超声:胆囊充盈欠佳,胆囊壁毛糙,目前未探及腹水。胸片:心、肺、膈未见明显异常。心电图:窦性心律,正常心电图。
为进一步明确诊断,行肝穿刺活检病理检查。
镜下所见:肝穿刺组织一条,小叶结构存在,II、III带肝细胞及毛细胆管明显淤胆,可见多核肝细胞,中央静脉周围少数小坏死灶。可见中小汇管区10个,部分汇管区轻度扩大,少量汇管区内混合性炎细胞浸润。免疫组化:HBsAg(-),HBcAg(-),CK7(胆管+),CK19(胆管+),MUM-1(散在细胞+)
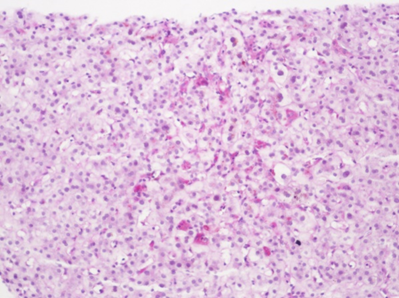
A 小叶中心胆汁淤积 D-PAS染色×200
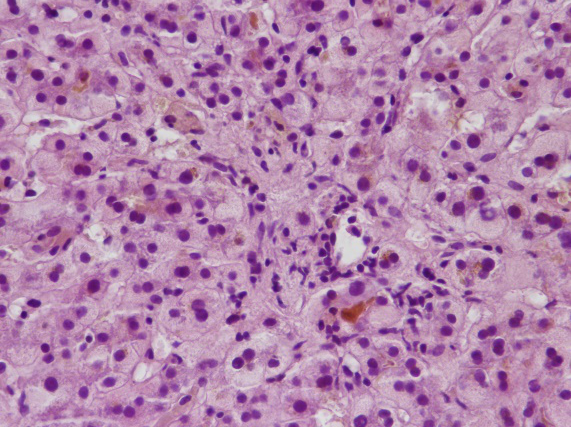
B 肝细胞内淤胆 HE染色×400
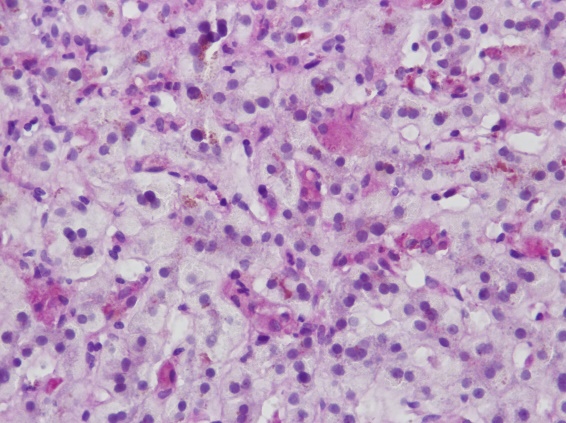
C 肝窦Kupffer细胞吞噬胆栓D-PAS染色×400
病理诊断:(肝穿)单纯性肝内胆汁淤积,请结合临床除外药物性淤胆或良性复发性肝内胆汁淤积及其他。
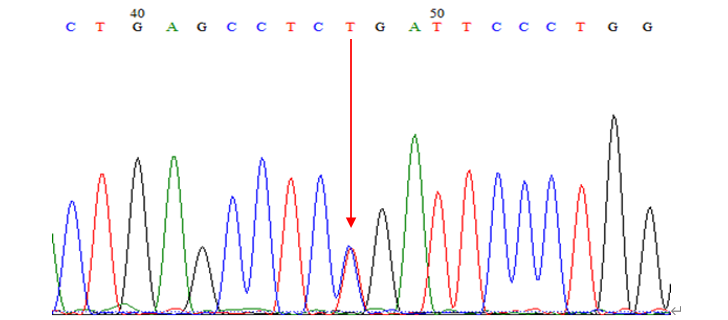
患者基因检测结果:检测到ATP8B1基因致病性突变(3040C>T,杂合突变,无义突变)
诊断:良性复发性肝内胆汁淤积症(BRIC1)
治疗:给予保肝、降酶及退黄对症支持等综合治疗,患者肝功能好转,病情恢复。
1.2 专家点评
张缭云教授: 该患者主诉“间断肝功能异常3年余”,从病史描述和检查结果看,实为胆汁淤积表现:皮肤发黄、瘙痒,反复胆红素明显升高,且以直接胆红素升高为主,ALP、TBA明显升高。首先应除外梗阻性黄疸。患者腹部超声:“胆囊充盈欠佳,胆囊壁毛糙,未见肝内胆管扩张”,所以定位缩小范围为肝内胆汁淤积。
进一步检查:各型病毒指标都阴性,自身免疫性肝病指标未见异常,凝血系列也正常,也无药物、嗜酒及环境毒物摄入和接触史,结合患者发病年龄偏年轻,所以应想到遗传代谢性肝病的可能。
肝穿刺病理可见:肝小叶结构存在,II、III带肝细胞及毛细胆管明显淤胆,与生化检查相呼应,主要病变是胆汁淤积。
进一步基因检测,结果发现ATP8B1基因存在致病性突变(3040C>T,杂合突变,无义突变)。综合患者临床表现及病理改变,患者确诊为良性复发性肝内胆汁淤积症(BRIC 1型)。
该病例应注意以下鉴别诊断:
1.对于黄疸明显升高患者,是肝细胞大片坏死还是胆汁淤积所致?
该患者肝酶基本正常,尤其凝血系列都正常,精神、食欲无明显表现,故肝衰竭可除外。
血清GGT正常的胆汁淤积症更多见于遗传代谢性疾病,应当足够警惕。这是因为GGT正常情况下分布于胆管上皮管腔的浆膜侧,在大多数胆汁淤积时(例如梗阻等),胆管腔内胆汁酸增多,有利于GGT从毛细胆管膜上洗脱下来,入血后引起GGT升高。当胆汁酸合成和运输障碍时(例如ATP8B1基因遗传缺陷),胆管腔内胆汁酸缺少或相对不足,故GGT可正常或低水平。
2.根据基因检测结果,应和ATP8B1基因突变的遗传代谢性肝病鉴别。
ATP8B1基因突变可导致包括进行性家族性肝内胆汁淤积症1型(PFIC1)、良性复发性肝内胆汁淤积症1型(BRIC1)和妊娠期肝内胆汁淤积1型(ICP1),可以统称ATP8B1基因缺陷病。ATP8B1基因缺陷病主要表现为血清GGT水平不升高的原因未明的胆汁淤积性肝病。
其中PFIC1以进行性胆汁淤积、黄疸伴瘙痒为特征,通常在1岁之前发病,平均发病年龄是3月龄。患者可在儿童早期迅速进展到终末期肝病,也可在十几岁时缓慢进展为肝硬化。患儿腹泻、营养物质吸收障碍及生长发育障碍较常见。肝外临床表现包括复发性胰腺炎、 腹泻、感音神经性听力损失、慢性咳嗽或喘息等。
ATP8B1基因突变的杂合子或者对蛋白功能损害较轻的突变的纯合子可表现为BRIC1。该病可反复发作胆汁淤积,自发缓解,不遗留严重的肝脏损害,发作间期无症状,首次黄疸可出现于1岁~50岁,多数在20岁之前发病,通常会有2~4周 的以乏力、食欲减退及瘙痒为特征的黄疸前期,但无明显的发作诱因,黄疸期可持续1~18月不等,以2~3月常见。在黄疸期,患者常出现体重下降及脂肪泻,生化检查可见血清胆汁酸、 胆红素及碱性磷酸酶水平升高,而GGT水平正常。肝组织活检提示良性改变。在无症状期,该病患者的临床表现、生化指标及肝组织病理均无异常。
ICP1可发生于PFIC1或BRIC1家系中,该病以孕期瘙痒和血清胆汁酸水平升高为特征,可导致早产、胎儿窘迫等严重后果。部分女性PFIC或BRIC患者可以ICP为首发表现。
本例患者20岁左右首次出现黄疸,以直接胆红素升高为主,GGT正常,治疗后胆红素可以降至正常,发作间期无特殊表现。反复发作,肝脏病理主要表现为胆汁淤积,未见肝细胞大片坏死及肝纤维化和肝硬化表现。基因检测发现ATP8B1基因致病性突变。综上,良性复发性肝内胆汁淤积1型(BRIC1)诊断明确。
有益提示:BFIC的诊断需要结合家族史、临床症状及体征、生化检查、影像学及病理学检查以及基因检测来综合判断。对于一些通过普通生化、影像甚至肝组织学不能确诊的病例,可借助基因检测来帮助诊断。在临床实践中,也发现人体是一个复杂的机体,同样的突变位点,受环境等外部因素影响,部分患者表现为BRIC,部分表现为PFIC,甚至个别患者由BRIC进展至PFIC。基因型和表型之间的关系也需要进一步研究。
1.3 病例供稿及点评专家简介
1.3.1供稿专家:

李璐:博士,主治医师
首都医科大学附属北京佑安医院疑难肝病及人工肝中心
1.3.2 点评专家:张缭云教授
2.文献分享
2.1文献标题及链接:ATP8B1缺陷病
2.2文献提供专家:王建设教授

王建设:博士,主任医师
复旦大学附属儿科医院感染传染科科主任
佑安专科联盟遗传代谢性肝病专业委员会顾问
四、专业委员会名单(按拼音排序):
白洁、白丽、包双宝、边巴央珍、卞丹丹、曾惜秋、常乐、陈芳、陈连清、陈琳、陈悦、程孟怀、程全红、次仁、代东旺、邓雪梅、丁建强、丁向春、杜方雄、段宏宪、段雪飞、段钟平、冯铁柱、高珍、葛迎春、巩维进、苟卫、顾伟玲、郭宁、郭文征、郭小青、何云、胡善雷、黄明星、黄祖雄、霍丽亚、江守伟、焦洪波、经继生、鞠莹、孔明、李宝生、李博、李灿、李广明、李红玲、李建国、李娟、李军、李俊峰、李磊、李秋莲、李荣宽、李响、刘彩峰、刘菲菲、刘晖、刘梅、刘霜、刘冉、刘晓彦、刘耀敏、刘振中、罗磊、吕帅、马玉秀、马臻、苗艳艳、牟丹蕾、南月敏、宁寒冰、牛卫理、彭鹏、乔晓红、邵鸣、盛云建、宋正已、苏丽、孙美艳、覃丽华、谭林、王德步、王飞、王健、王建设、王丽华、王小凤、王艳巧、王仲培、吴刚、吴万锋、吴晓枫、向光明、肖玉珍、谢秋里、易永芬、杨艳玲、于德顺、袁喜先、张定琳、张帆、张立婷、张丽、张丽娟、张缭云、张妍、张银华、张玉山、张月荣、张志刚、张宗超、赵守松、赵素贤、郑素军、周晓丽、周晓玲、邹桂舟
五、联系方式
□投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
□联系电话:010-63291007
□联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间