
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:钟雪梅
执行编辑:郑素军,汤珊,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
希特林蛋白缺陷病(Citrin Deficiency,CD)是一种常染色体隐性遗传疾病,citrin蛋白由SLC25A13基因编码,是一种钙结合溶质载体蛋白,主要表达于肝细胞线粒体内膜,作为天冬氨酸、谷氨酸载体将线粒体内的天冬氨酸转运至胞质,同时将胞质的谷氨酸和质子转运至线粒体内部,在能量、氨基酸、糖和脂肪代谢中发挥重要作用。肝细胞能量缺乏是本病的基本发病机制。
目前已报道的 SLC25A13 基因变异有100 余种,突变类型包括缺失突变、错义突变、无义突变及剪接位点突变。基因突变在我国以c.851_854del、c.IVS6+5G>A和 IVS16ins3kb最常见,其中发生频率最高的是c.851_854del。北方群体的 SLC25A13 等位基因异质性明显高于南方群体。虽然 SLC25A13 基因突变分布有明显的种族及地域差异,但与患儿的临床表现、异常生化指标无明显关联性。
CD有三种年龄依赖性临床表型:citrin缺陷导致的新生儿肝内胆汁淤积症(Neonatal intrahepatic cholestasis caused by citrin deficiency,NICCD)、citrin缺陷导致的发育不良和血脂异常(Failure to thrive and dyslipidemia caused by citrin deficiency, FTTDCD)、成人发病的瓜氨酸血症II型(Adult-onset type citrullinemia Ⅱ, CTLN2)。CD于婴儿期起病,临床表现复杂多变,早期症状常可自行缓解,但年龄较大的患者可能出现严重的合并症。儿科、消化科、肝病科、神经内科医生都需要深入了解此病。
NICCD是最为常见的类型,患者多于2月龄内就诊,表现为新生儿黄疸延迟消退、胆汁淤积、肝功能异常、肝脾肿大、多种代谢异常。其治疗主要是饮食疗法,无乳糖、强化中链脂肪酸(MCT, Medium chain Triglycerides)特殊配方奶粉的疗效已得到证实。NICCD患者大多数在1岁以后临床症状消失,但是也有在婴儿期因发生肝衰竭需要进行肝移植的报道。CTLN2多在青春期后发病,一般是由于乙醇和糖摄入过多或者手术诱发,高氨血症导致的神经精神症状为突出临床表现,往往预后不良。补充精氨酸及饮食疗法能够减轻患者症状,病情严重的患者需接受肝移植。2011年我国学者提出FTTDCD为NICCD和CTLN2的过渡型,其临床表现多样且缺乏特异性,且并非所有患者都有NICCD病史。FTTDCD患者喜欢高蛋白和高脂肪的食物,厌恶乙醇和碳水化合物,往往体型偏瘦、生长迟缓、易疲劳,可出现低血糖、血脂异常、胰腺炎、脂肪肝、肝细胞瘤等。高脂肪、高蛋白、低碳水化合物的饮食疗法是年长儿最重要的治疗方法,由于CD患者的能量消耗高于普通人群,因此患者每日总能量摄入应高于常规摄入量,并且推荐蛋白:脂肪:碳水化合物比例为15 - 25%:40-50%:30-40%。
本期月报报道1例希特林蛋白缺陷病合并胆管结石、胰腺炎、胆总管囊肿的临床病例。该病例婴儿期以皮肤黄染为主要表现,肝功能检查异常,经基因检测后最终诊断明确并给予饮食治疗,患者病情控制良好,生长发育指标正常。2年后(患儿2岁8月龄)出现胆管结石、急性胰腺炎,进行了ERCP+EST+EPBD+球囊取石术,术后胰腺炎反复发作,最终诊断为胆总管囊肿,接受Roux-y手术后胰腺炎治愈。通过对该病例的分析,希望临床医生提高对希特林蛋白缺陷病的认识,对于原因不明的黄疸、肝功能损害,需要积极进行相关基因检测,警惕遗传代谢疾病等罕见病存在;同时对于需要特殊饮食治疗的疾病需要加强饮食指导以及长期随访。

钟雪梅
首都儿科研究所附属儿童医院消化内科 主任医师
中国医师协会儿科医师分会委员
中华医学会消化病学分会儿科协作组委员
中华医学会肝病学分会遗传代谢性肝病协作组委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1. 线粒体天冬氨酸/谷氨酸载体的致病性变异导致的希特林蛋白缺陷病(Trends in Endocrinology & Metabolism,2022, IF=10.536; Q1区)

2. 生酮饮食治疗线粒体苹果酸天冬氨酸通道和丙酮酸载体的缺陷(Nutrients,2022, IF=6.706; Q1区)
3.希特林蛋白缺陷病导致的新生儿肝内胆汁淤积症代谢特征的动态变化(Frontiers in Molecular Biosciences,2022, IF=6.113; Q1区)
4.希特林蛋白缺陷病的临床表现和长期预后:一项来自日本的全国性研究报告(J Inherit Metab Dis,2022, IF=4.75; Q2区)
5.希特林蛋白缺陷病患者每日能量、蛋白质、脂肪和碳水化合物摄入分析(Molecular Genetics and Metabolism,2022, IF=4.204; Q2区)
6.低血糖发作和生长衰竭是希特林蛋白缺陷病1岁后最常见的表现(J Inherit Metab Dis,2021, IF=4.75; Q2区)
7.希特林蛋白缺陷病患者的食物偏好(Nutrients,2021, IF=6.706; Q1区)
8.血清尿素氮或尿素氮/肌酐可能作为希特林蛋白缺陷病标志物(Molecular Genetics and Metabolism Reports,2022, IF=2.082; Q4区)
9.希特林蛋白缺陷病所致的新生儿肝内胆汁淤积症不伴脂肪变性:病例报告(BMC Pediatrics,2021, IF=2.567; Q3区)
10.综述:希特林蛋白缺陷病的代谢基础及治疗(J Inherit Metab Dis,2021, IF=4.75; Q2区)
三、临床资讯
1.1 病例分享
第一次住院(2015年11月)
现病史
男,6月5天,因“发现皮肤巩膜黄染6月”于2015年11月03日入院。入院前6月(即生后5天)发现患儿皮肤、双眼出现黄染,尿色不深,大便为黄色糊便,4-5次/天,无发热、呛咳、吐奶等不适,未予特殊诊治;入院前4月余(即生后42天),皮肤巩膜黄染无消退,经皮测胆红素12mg/dl,停母乳,加用茵栀黄对症口服一周,皮肤黄染较前加重,尿色深,大便为黄色糊便,无白陶土色便,当地医院完善生化示转氨酶正常,胆红素升高,直接胆红素为主,病因不明,故转我院进一步诊治。
生长发育史
3个月抬头,4月余翻身,目前6个月,可扶坐。
孕产史及家族史
患儿系第一胎第一产,足月顺产,出生体重3.6 kg。母亲孕期体健,家族中无类似疾病患者。
体格检查
体重8kg,身长68cm,神清,反应好,面色正常,皮肤巩膜明显黄染,双肺呼吸音清晰,呼吸平稳,无发绀,无三凹征,心律齐,心音有力,未闻及杂音,腹部稍膨隆,肝脏肋下100px,质韧,边钝,脾肋下未及,肠鸣音正常,神经系统查体未见异常,末梢暖。
辅助检查
①血生化:ALT 47.9U/L,AST 105.8U/L,γ-GGT 78.3U/L,TBIL 119.3umol/L,DBIL 24.6umo1/L、TBA 516umol/L;胆固醇 3.21mmo/l,甘油三酯 2.68mmo/l,低密度脂蛋白 2.83mmo/l,血糖 5.66mmol/l;血氨 62umol/l;
②凝血:凝血酶原时间 不凝,活化部分凝血活酶时间 85.3s,INR 不凝,纤维蛋白原 1.4g/l,D-二聚体 45ug/l;
③TORCH抗体阴性;CMV-DNA阴性;EB-DNA阴性;
④甲功未见异常;
⑤甲胎蛋白>2000ug/ml;
⑥腹部超声:左侧肾盂肾盏增宽,肝胆胰脾未见明显异常;
⑦血氨基酸分析:瓜氨酸/苯丙氨酸、蛋氨酸/苯丙氨酸、长链脂肪酸亚油酸(C18:2)增高;谷氨酰胺、短链脂肪酸初油酸(C3)降低;
⑧尿液有机酸分析:蛋氨酸、腺嘌呤、半乳糖和腺苷增高。
基因检测
行遗传代谢性肝病二代基因panel测序,检测发现SLC25A13基因2个突变位点:①cc.852_855delCATA,为移码突变,来源于其父亲;②c.550G>A,为错义突变,突变来源于患儿母亲。
治疗及随访
根据患儿血生化、血尿代谢筛查、基因检测等检查,诊断citrin缺陷导致的新生儿肝内胆汁淤积症(NICCD)明确。予保肝退黄等治疗及无乳糖、强化中链脂肪酸配方粉喂养,并予积极补充脂溶性维生素。
患儿10月龄时皮肤黄染好转,1岁后予小百肽及高蛋白饮食喂养。2岁5月龄因“传染性单核细胞增多症”于我院就诊,期间完善血生化示ALT 63.8U/L,AST 42U/L,胆红素、胆汁酸均正常,对症抗病毒、营养脏器治疗后转氨酶降至正常。
第二次住院(2017年12月)
现病史
患儿2岁8月龄,因“腹痛3天”入院。入院前3天患儿进餐后出现腹痛,伴哭闹,不易安抚,持续约半小时至1小时后逐渐缓解,无发热、呕吐等不适,无明显皮肤巩膜黄染,遂就诊于我院急诊查血常规:WBG 9.49×109/L,N 59.1%,L 34.2%,M 6.5%,HB 124G/L,FLT 299×109/L,CRP 7mg/L,血生化:ALT 107.2U/L,AST 90.9U/L,血淀粉酶727U/L,总胆红素17.9umo1/L,直接胆红素4.4umo1/L,电解质未见异常;甘油三酯 2.56mmol/L,总胆固醇、脂蛋白正常。遂为进一步诊治以“急性胰腺炎”收入院。
辅助检查
①腹部增强CT:胰腺形态及密度尚正常,胰头及胰体近侧似见纤细胰管影。肝内胆管及胆总管扩张。肝周及胆囊窝内少量积液;
②腹部超声:胆囊管、胆总管、以及肝内胆管左右支均可见扩张,肝内胆管最宽12.5mm,肝外胆管最宽11.1mm,腔内可见多个大小不等的中-强回声团影;于胆总管末端胰胆合流下方可见一较大中强回声影,约6.7×6.7mm,胆囊壁增厚,回声减低,胰腺回声增强,未见明显肿大,胰管宽约2.4mm。
治疗
入院后予禁食水,对症补液,拉氧头孢及奥硝唑静点抗感染,复合辅酶、复方甘草酸苷营养脏器对症保肝治疗,思美泰静点、熊去氧胆酸胶囊口服促进胆汁排泄,奥美拉唑静点抑酸;入院后第16天患儿行ERCP+EST+EPBD+球囊取石及胃空肠营养管置入术检查,术后奥曲肽连续72小时泵维预防术后胰腺炎,小百肽稀释奶泵维,并逐渐由泵奶过度至经口喂养,入院后第29天患儿经口喂养良好,好转出院。
出院后20余天胰腺炎反复,复查腹部B超:胆总管扩张,5.4mm,内可见中强回声,左右肝管直径5.7mm,胰管未见扩张。在全麻下行腹腔镜探查术+胆管造影术,术中见胆总管梭形扩张,胰胆共同管直径5mm,长50px,胰管显示不清,肝内胆管扩张,遂行Roux-y手术,后胰腺炎治愈。
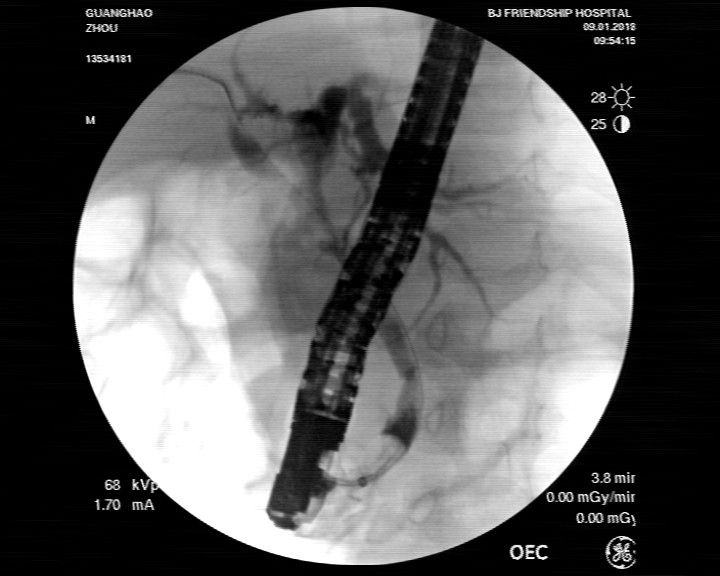
图1:ERCP示胆总管扩张,胆总管下段见结石影
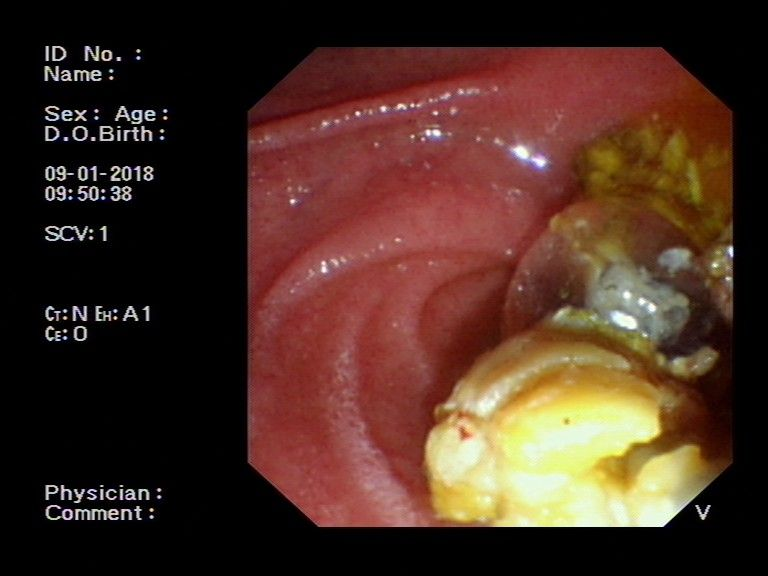
图2:切开十二指肠乳头后,以扩张球囊逐级扩张乳头开口,取石球囊取出结石
1.2供稿专家简介

马昕
首都儿科研究所附属儿童医院消化内科 主治医师

刘文雯
首都儿科研究所附属儿童医院消化内科 主治医师
四、联系方式
●投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
●联系电话:010-63291007
●联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间