
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张岭漪
执行编辑:郑素军,梁晨,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
范科尼贫血(fanconi anemia,FA)是最常见的遗传性骨髓衰竭性综合征(heritageed bone marrow failure syndrome,IBMFS),是染色体不稳定综合征的主要类型之一。在不同种族、不同地区发病率有所不同。在亚洲人群中发病率为1/160, 000,男女发病比例约1.2:1。除FANCB-相关FA为X连锁遗传,RAD51-相关FA为常染色体显性遗传外,其余FA为常染色体隐性遗传。目前已发现至少22个FA基因突变,最常见的突变分别为FANCA(65%)、FANCC(15%)、FANCG(10%)、FANCE和FANCF(8%),其他各种基因突变类型<1%。FA的发生是由于编码FANC/BRCA通路蛋白的基因突变,这些基因通过断裂诱导复制(BIR)或同源重组(HR)管理单端或双端双链断裂(DSB)修复,参与DNA链间交联(ICL)修复和复制挽救。FANCA纯合子缺失突变可能早期出现贫血,白血病发病率高于其他能产生异常FANCA蛋白的致病突变。
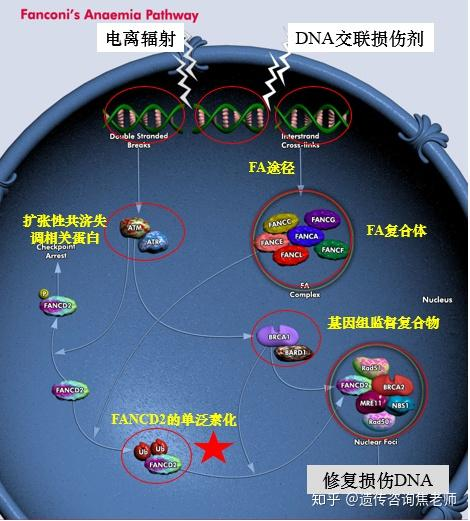
不同基因突变或缺失可表现为不同的亚型和临床表现。FA主要临床表现为先天性发育异常、进展性骨髓衰竭和肿瘤发生率增高,也有患者无发育异常或骨髓异常表现。发育异常的临床表现多种多样,如:骨骼畸形、皮肤色素沉着、生长缺陷、泌尿生殖道畸形及心血管系统发育异常。进展性骨髓衰竭则多表现为血细胞减少。中位发病年龄约为7.6岁,婴儿和幼童罕见,90%患者的血液学异常在40岁前发病。血小板减少或白细胞减少通常先于贫血出现,常伴随大红细胞、胎儿血红蛋白增加,全血细胞减少逐渐加重。血液异常进展可出现Sweet综合征(中性粒细胞皮肤浸润)。FA是遗传性肿瘤易感综合征,急性髓系白血病(AML)发生危险增加约700倍,未行移植治疗的FA患者40岁时约有50%发生骨髓增生异常肿瘤(MDS)或AML。多发生于1535岁之间。实体肿瘤可以是FA的首发表现。头颈部鳞状细胞癌(HNSCCs)是FA患者最常见的实体瘤,较普通人群发病率增加500~700倍,而发病年龄更早(20~40 岁),大多数发生于口腔,对治疗反应差。皮肤肿瘤、食管癌、肝癌和泌尿生殖道鳞状细胞癌发生率亦增加。最后,亦偶有患者可以肝脏异常为首发表现。
FA的诊断仍然基于临床表现、染色体断裂增加(尤其是暴露于DNA链间交联剂)和基因异常,如合并肝脏异常,必要时需行肝组织穿刺活检。FA的治疗方面无论是《2014年共识会议商定的范科尼贫血治疗意见》还是最新的《范科尼贫血诊断与治疗中国专家共识(2022版)》均无绝对有效的治疗推荐,药物治疗包括使用雄激素纠正贫血,如患者骨髓衰竭进行性恶化,仍应考虑进行骨髓移植治疗。FA患者的DNA修复缺陷使患者对实体瘤的放化疗都敏感,因毒性增加可能出现致死性并发症,因此实体瘤的治疗仍有赖于早期发现、手术切除治疗。但对于晚期无法手术切除的患者,仍需采取放疗或者化疗治疗,或者与手术结合治疗,尽管目前还没有明确的剂量减量方法。多年来,有专家研究FA的基因治疗,虽然仍处于体外矫正基因缺陷的阶段,但基因治疗是一种未来有望替代异基因造血干细胞移植治疗FA的新型低毒性治疗方式。
FA作为一种罕见的单基因缺陷疾病,其遗传方式包括常染色体隐性遗传、常染色体显性遗传(RAD51-相关FA),或X连锁遗传(FANCB-相关FA)。因此,如果已知家族致病基因的情况下,常染色体隐性遗传和X连锁遗传FA的亲属均应做携带者检测和妊娠产前检测。
就本例患者,我们亦不难看出,本患者以突出的皮肤着色异常及肝损为首发临床表现,同时出现进行性加重的骨髓衰竭综合征,因此极易被非血液专科医生误诊及漏诊。同时,FA合并肝损的患者在临床中亦十分罕见。其二,本例患者因就诊时全血细胞已属重度减少,同时凝血功能亦受到影响,存在肝脏组织活检禁忌症,因此未能取到肝脏组织标本。但依然可根据患者突出的临床表现以及全外显子基因测序,最终确诊为范科尼贫血互补群A。其三,目前该患者尚未出现肿瘤的表现,但因影像学中已多次诊断肝脏异常结节,因此今后仍需密切随访。最后,患者的两个儿子为隐性携带者,分别携带一个FANCA基因杂合突变位点,因此其后代是否会发病尚不可知,需做好其配偶的检测和筛查。

张岭漪
主任医师,教授,研究生导师
兰州大学第二医院肝病科 主任
中华医学会肝病学分会、预防感染病防控分会委员
中国医疗保健国际交流促进会、肝胆疾病学分会常务委员
中国医药生物技术协会慢病管理分会常务委员
中华医学会肝病学分会肝纤维化学组委员、终末肝学组委员
甘肃省医学会肝病专业委员会主任委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.范科尼贫血患者的基因型表型和结果关联:美国国家癌症研究所队列(Haematologica,2023,IF=10.10; Q1区)

2.中国罕见病队列中148名范科尼贫血儿患者的表型和基因型相关性评价(Clinica Chimica Acta,2023,IF=5.00; Q1区)
3.范科尼成年患者的表型(British journal of haematology,2023,IF=6.50; Q1区)
4.遗传性癌症综合征的评估强调范可尼贫血为口腔潜在恶性疾病(International journal of surgery,2023,IF=15.30; Q1区)
5.识别范科尼贫血中的强大DNA甲基化特征(American journal of human genetics,2023,IF=9.80; Q1区)
6.基因治疗恢复范科尼贫血造血干细胞的转录程序(Haematologica,2023,IF=10.10; Q1区)
7.通过基因转换事件逆转表型作为范科尼贫血的“自然基因疗法”(Frontiers in genetics,2023, IF=3.70; Q2区)
8.综述:范科尼贫血基于细胞和基因治疗方法进展(Blood reviews,2023,IF=7.40; Q1区)
9.先天性角化不良的肝脏病理改变(The American journal of surgical pathology,2023,IF=5.60; Q1区)
10.先天性角化不良及相关的端粒生物学障碍中肝脏疾病和门静脉高压的进展(Hepatology,2023,IF=13.50; Q1区)
三、临床资讯
3.1 病例分享:1例范科尼贫血病例
患者男性,38岁,间断乏力8月余。
现病史:患者于入院前8月,无明显诱因出现间断乏力,伴有间断发热,体温最高为41℃,伴有胸闷、气短,腹胀,但无腹痛,无恶心、呕吐,无呕血、黑便,遂就诊于我省某三级医院,门诊行相关检查(具体不详)后考虑为“肝硬化”,建议患者住院治疗,但予以拒绝,遂给予“谷胱甘肽 0.4g tid,葡醛内酯 0.2g tid”治疗,后症状略有缓解。于本次入院前2周,患者无明显诱因再次出现乏力症状,并伴有胸闷、气短,就诊于空军军医大学第一附属医院,门诊完善相关检查。建议住院治疗,但患者因个人原因,予以拒绝,给予“熊去氧胆酸 250mg tid,甲钴胺 500µg tid,叶酸片 5mg tid”。但乏力症状无缓解,遂患者为进一步明确诊治,就诊于我院门诊,并收住入院。
既往史及家族史:患者于2年前于当地医院诊断“白癜风”,但未给予任何药物治疗。否认手术外伤史,否认传染病及其接触史,否认药物过敏史,否认输血史。否认吸烟、饮酒等不良嗜好。否认药物嗜好,否认毒物粉尘接触,否认冶游史。患者已婚,育有2子(大儿子12岁,小儿子9岁)。否认家族相关症状及遗传病史。其父母否认近亲结婚。
入院查体:神志清,精神尚可,慢病面容,巩膜轻度黄染,眼睑明显苍白。颜面皮肤呈斑片状咖啡样色素脱失(见图1)。双手、双上肢、前胸以及后背均可见斑片状色素脱失,类似于“白癜风”皮肤表现(见图2)。双肺底呼吸音减低,以右侧为著,未闻及干湿性啰音。腹部略膨隆,腹软,全腹无压痛及反跳痛,脾肋下3横指可触及,质硬、活动度差,移动性浊音可疑阳性。
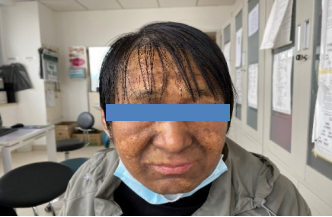
图1
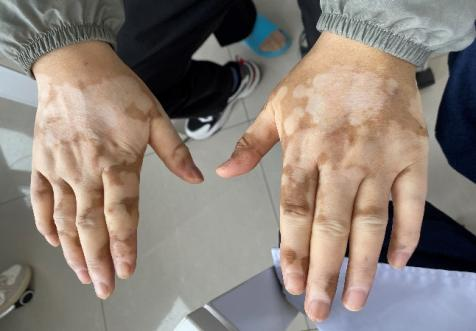
图2
一般检查:血常规:白细胞 0.76×109/L,血红蛋白 50g/L,血小板 23×109/L;生化:总胆红素 54.9µmol/L,直接胆红素 18.6µmol/L,ALT 22U/L,AST 31U/L,白蛋白 36.2g/L,碱性磷酸酶 255U/L,GGT 40U/L,乳酸脱氢酶 396U/L,总胆汁酸 70.3µmol/L;凝血:PT% 53%,FIB 1.72g/L;铁蛋白:514ng/mL;铜蓝蛋白 45.7mg/dL(正常范围);嗜肝病毒阴性,自身抗体检查:ANA(弱+),抗平滑肌抗体(+),AMA-M2阴性;Fibroscan:13.7Kpa,ICG-R15:8.9%;抽取胸腔积液,胸水常规:Rivalta 试验(弱+),白细胞 369×106/L。
影像学检查:彩超提示:肝硬化,肝右叶可疑实性结节,门静脉略增宽,脾大并脾静脉增宽,腹腔少量积液,右侧胸腔积液(中量),左侧胸腔积液(少量);腹部平扫:肝硬化并S7再生结节形成、门脉高压-侧枝循环形成、脾大、腹腔及右侧胸腔积液;普美显核磁检查:1. 肝S8 DWI稍高信号,未见强化;2.肝实质信号欠均匀,T2WI信号明显减低,考虑血色病可能;3.肝胆期肝实质弥漫性信号减低,考虑肝脏储备功能减低;肝病胆囊。
骨髓穿刺与活检:骨髓穿刺:骨髓增生明显活跃,粒红比值倒置,粒系及巨核系可见成熟障碍。血象:白细胞明显减少,以中性粒及淋巴细胞为主,可见胞浆颗粒增多;成熟红细胞大小不等,有中空,可见异形红细胞;血小板少呈散在分布。骨髓活检:造血组织增生活跃,粒系以中晚幼及以上阶段为主,幼红细胞比例增高,巨核细胞偏少。
本次住院期间,虽然患者处于重度贫血状态,但因铁蛋白升高明显,同时亦不能完全排外血色病,因此未予患者输注红细胞纠正贫血。通过抽取胸水,以及基础保肝治疗后,患者胸闷、气短症状较入院时有所缓解。结合患者影像学及相关检验结果,该患者仍有众多疑点,例如:肝硬化诊断是否成立?如果成立,能否解释三系减低,尤其是重度贫血?患者皮肤表现又如何理解?一元论?二元论?此时,鉴于以上疑问,完善全外显子基因测序 。
全外显子基因测序:该案例,先证者在FANCA基因c.1776-92G>C(染色体位置:chr16:89845259)发现一处杂合突变,父亲为野生型,大儿子为杂合突变,小儿子为野生型,ACMG评级为PM2+PM3+PP3+PP4,可能致病;在FANCA基因c.4096C>T(染色体位置:chr16:89805612)发现一处杂合突变,父亲为杂合突变,大儿子为野生型,小儿子为杂合突变,ACMG评级为PVS1+PM2,可能致病。经家系验证表明两处突变分别位于不同染色体,构成复合杂合突变,符合疾病“范科尼贫血互补群A”隐性遗传规律。(见图3)
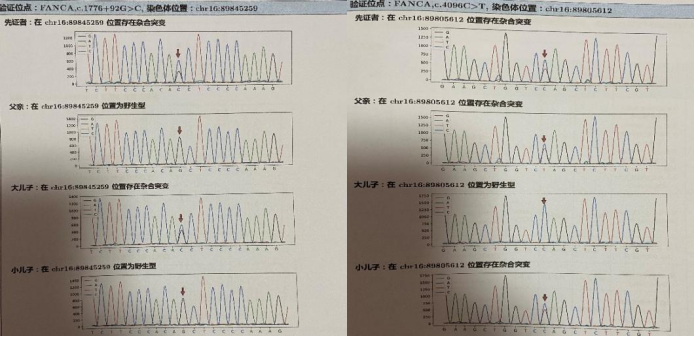
图3
诊断:患者为青年男性,病史有近2年的时间,突出的临床表现有:⑴皮肤着色异常:咖啡斑、瘀斑易感性、贫血性白斑,且一度诊断为“白癜风”;⑵黄疸、肝损:胆红素升高、脾脏肿大;⑶全血细胞减少;⑷纠正贫血治疗无效。并结合骨髓穿刺以及全外显子基因测序可明确诊断。本例患者因就诊时全血细胞已属重度减少,同时凝血功能亦受到影响,存在肝脏组织活检禁忌症,因此未能取到肝脏组织标本。但从患者突出的临床表现以及全外显子基因测序,可确诊为范科尼贫血互补群A。
治疗:参照2014年共识会议商定的范科尼贫血治疗意见,雄激素能改善(至少短暂地)大约50%的患者中红细胞和血小板计数。当血红蛋白降至8 g/dL以下或血小板计数低于30,000/mm3时,可考虑使用雄激素治疗。虽然只有10%-20%接受持续低剂量雄激素治疗的患者有长期效果,选择雄激素对部分无法进行造血干细胞移植(HSCT)或者没有合适捐髓者的患者有效。因此,给予患者口服“达纳唑”治疗。如患者骨髓衰竭进行性恶化,仍应考虑进行骨髓移植治疗。近期电话随访该患者,目前仍在小剂量服用雄激素治疗,病情尚平稳。肝功能较前无明显变化,白细胞水平略有回升,但由于达那唑的副作用较大,无法足量给药,治疗效果尚不显著。
最后,幸运的是患者的两个儿子为隐性携带者,分别携带一个FANCA基因杂合突变位点,因此其后代是否会发病尚不可知。
3.2 供稿专家简介
张岭漪
主任医师,教授,研究生导师
兰州大学第二医院肝病科 主任
中华医学会肝病学分会、预防感染病防控分会委员
中国医疗保健国际交流促进会、肝胆疾病学分会常务委员
中国医药生物技术协会慢病管理分会常务委员
中华医学会肝病学分会肝纤维化学组委员、终末肝学组委员
甘肃省医学会肝病专业委员会主任委员

何晶晶
博士,副主任医师
中国医促会肝脏肿瘤分会青年委员
中华预防医学会感染性疾病防控分会青年委员
全国肝健康促进专家委员会委员
甘肃省医学会肝病学分会青年委员
甘肃省医学会感染病学分会青年委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间