
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张国民
执行编辑:郑素军,梁晨,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
肝豆状核变性(Wilson disease,WD)为一种常染色体隐性遗传代谢性疾病,表现为铜代谢障碍,因其患病率较低(1/30 000),被列入罕见病目录,但在部分亚洲国家其发病率却可高达 1/5 400。目前已知ATP7B是WD的致病基因,其主要编码一种铜转运P型蛋白ATP酶,该酶的功能异常可引起铜蓝蛋白减少及胆道排铜障碍,进而过量的铜沉积导致肝、脑、骨骼等组织器官损伤。WD表现异常多变,仅肝脏病理就包括从轻度炎症、明显脂肪变性、肝硬化到突然肝衰竭。该病早期起病较为隐匿,缺乏特异性,常常被误诊、漏诊,而及早识别 WD 有利于规范治疗和改善预后。
非酒精性脂肪性肝病(non-alcoholic fatty liver disease, NAFLD)是一种与胰岛素抵抗和遗传因素密切相关的代谢应激性肝损伤,包括非酒精性肝脂肪变、非酒精性脂肪性肝炎、肝硬化和肝细胞癌。该病虽然常见于肥胖患者,但也可能由其他病因引起,例如WD。现已有多例报道显示一些肥胖患者同时存在NAFLD和WD;同时亦发现,肝周脂肪层可以是WD影像学特有表现,WD患者肝脏可显示与脂肪性肝炎类似的病理改变,如肝细胞脂肪变性、气球样变,甚至脂肪变性可以是其唯一的病理变化。这增加了WD患者可能会被误诊为NAFLD的机会,故在《非酒精性脂肪性肝病防治指南》中明确指出:即使肝组织学或影像学明确存在弥漫性脂肪肝表现,在归结于 NAFLD 之前,亦需要除外多种可同样导致脂肪肝的特定肝病,其中就包括WD。
那么,WD与肝脂肪变之间又有怎样的关系呢?一项关于WD患者组与健康对照组营养调查研究显示,与健康对照组相比,WD患者全身和躯干的脂肪量和脂肪率显著增加(P < 0.001),全身和躯干的肌肉和骨骼肌量显著减少(P < 0.001)。另有研究人员发现具有WD铜特征(即高肝铜、低血清铜和盲肠血浆蛋白)新生豚鼠的肝脏脂肪评分与肝铜浓度呈显著正相关(r=0.60;p < 0.001),其他关于WD的研究中,也同样证实肝铜较高时,脂肪变性似乎更明显,这提示铜潴留与肝脏脂肪变性的发生可能有关。目前WD患者肝脏损害和脂肪变性的机制仍然不清楚,有研究表明,过氧化物酶体增殖物激活受体(PPARs)γ和α的变化与WD肝脏脂肪变性和抗氧化系统损伤有关。有意思的是在NAFLD(包括非酒精性脂肪肝(NAFL)和非酒精性脂肪性肝炎)中观察到铜失衡,但是铜含量的变化与WD的情况相反。NAFLD患者肝内和血清铜是降低的,此外,肝脂肪和铜含量呈负相关。NAFLD中的铜缺乏和WD的铜超负荷是如何触发非常相似的肝脏代谢和形态学变化尚不清楚,仍有待进一步研究。
本期月报介绍的这例患者,WD患者合并重度肥胖,曾在发病早期被误诊为非酒精性脂肪肝,错过了早期的有效诊治,进而发展为肝硬化失代偿期,故对于影像学、病理学明确存在脂肪变表现的患者,特别是儿童及青年患者,需注意筛查WD的可能,做到早期筛查,早期发现,早期治疗,减少漏诊、误诊的发生,以期改善这部分患者预后,延缓其并发症的发生。

张国民
主任医师,硕士研究生导师
承德医学院附属医院感染控制处处长,感染性疾病科主任
全国疑难及重症肝病公关协作组第五届全国委员
全国肝健康促进专家委员会委员
从事感染性疾病的诊疗、教学、科研工作20余年
参编十三五规划教材《传染病学》两部
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.中国Wilson病ATP7B R778L突变:系统回顾和meta分析(Pediatric neurology,2023,IF=3.80; Q1区)

2.Letter to editor: Wilson病早期神经系统症状恶化的定义(Journal of hepatology,2023,IF=25.70; Q1区)
3.综述:Wilson病进行基因检测的诊断意义与困惑(Diagnostics,2023, IF=3.60; Q2区)
4.纯肝性Wilson病患者出现神经系统症状的危险因素(BMC neurology,2023,IF=2.60; Q3区)
5.Wilson病患者长期治疗中神经系统症状、肝功能异常恢复时间的差异(Journal of clinical medicine,2023,IF=3.90; Q2区)
6.Wilson病患者停用金属螯合剂治疗后症状加重(Brain and behavior,2023,IF=3.10; Q2区)
7.Wilson病患者深灰质核中的金属沉积与脑萎缩有关(Cerebral cortex,2023,IF=3.70; Q2区)
8.Wilson病基底神经节的显微结构和功能改变(Frontiers in neuroscience,2023,IF=4.30; Q2区)
9.西班牙Wilson病患者健康生活质量:一项横断面观察研究(Journal of clinical medicine,2023,IF=3.90; Q2区)
10.Wilson病tx-J动物模型海马组织编码和非编码RNA转录组表达谱的综合分析(Scientific reports,2023,IF=4.60; Q2区)
三、临床资讯
3.1 病例分享:1例表现为重度肥胖的肝豆状核变性患者
患者马某,男性,30岁,主因“腹胀20天,加重伴周身水肿6天”于2023年2月6日就诊。
现病史:患者于20天前无明显诱因出现腹胀,以剑突下为著,进食后加重,伴尿色加深,如茶色,尿量较前减少(具体量不详),无腹痛,无恶心、呕吐,无反酸、烧心,无呕血、黑便,无发热,无腰痛及肉眼血尿,间断口服“健胃消食片1片日三次”,上述症状无缓解。6天前患者腹胀加重,伴周身水肿,水肿由双下肢逐渐累及眼睑、颜面、双上肢,伴胸闷、气短,活动时为著,无端坐呼吸、胸痛、咯血,就诊于佛山市某医院,查腹部CT平扫:考虑肝硬化、脾大、腹水及胃底食道静脉曲张,肝左叶稍低密度影,腹膜后多发稍肿大淋巴结。考虑“肝硬化 脾大 腹水 胃底食道静脉曲张”,患者未求进一步诊治,故就诊于我院肝病门诊,门诊以“肝硬化”收入院。
既往史、个人史及家族史:患者生长发育无特殊。既往26年前患“淋巴结结核”,经治疗半年已治愈(具体治疗药物不详)。发现脂肪性肝病8年,未系统诊治。4个月前患者因右下肢水肿于佛山市某医院就诊,考虑“右下肢静脉曲张”,给予保守治疗(具体治疗不详)31天,水肿减轻后出院,院外未用药,仍间断水肿。“头孢类药物、阿莫西林”应用过敏史。BMI 52.7(重度肥胖);吸烟史12年,1年前戒烟,平均10支/日,近10年偶有少量饮酒。家族中母亲及妹妹肥胖,父亲体型正常,体健。
入院后完善检查:血常规:WBC 2.38×10^9/L,HGB 115g/L,PLT 72×10^9/L,N% 61.6%。肝肾功能:TBIL 34.53 µmol/L,DBIL 9.57µmol/L,GGT 34.8U/L,ALP 104.7U/L,ALT 33.6U/L,AST 53.6U/L,ALB 29.8g/L,CREA 44.3µmol/L;凝血项:PTA 60.3%,血脂:apoB 0.55g/L,余TC、TG、HDL-C、apoA I、LDL-C未见异常;血氨 41.6;嗜肝病毒检测(HAV、HBV、HCV、HEV、CMV、EBV)阴性,自身免疫抗体正常;AFP:正常。免疫球蛋白未见异常;心脏彩超:未见异常;腹水彩超:腹腔可见液性暗区,最大液深94.4mm;增强CT:肝硬化、脾大、食管胃底静脉曲张、腹腔积液。头颅磁共振:脑内多发缺血灶。血管彩超:脑血管多普勒超声,双侧颈动脉、颈内动脉、椎动脉,双侧锁骨下动脉超声均未见异常。
腹部CT
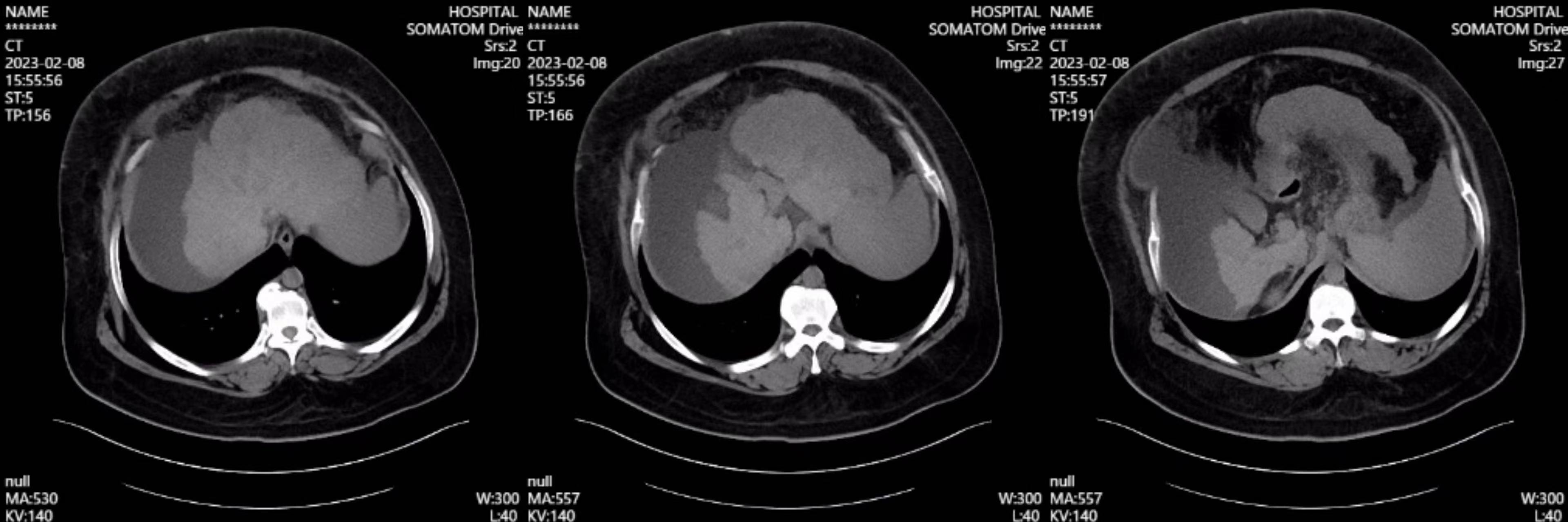
患者重度肥胖,曾明确诊断脂肪肝,临床医师曾一度考虑脂肪肝所致肝硬化,患者进食量不多,考虑肥胖病因不除外继发所致。
内分泌会诊:筛查皮质醇节律正常,不支持库欣综合征;甲状腺功能、甲状腺抗体正常,不支持甲状腺功能减退;葡萄糖耐量实验、空腹及服糖后血糖正常,胰岛素水平正常,不支持糖尿病;雌二醇高考虑不除外与肝病及肥胖有关。泌乳素略高于正常,但患者无溢乳等表现,嘱其定期复查。
治疗效果:经对症保肝、降酶、退黄、补充蛋白、利尿等治疗,效果差,白蛋白无改善(30g/L),凝血功能恶化(PTA 53%),腹水量增加(119mm)。肝硬化、腹水、血小板减低、凝血功能差,存在经皮肝穿禁忌。
后续结果回报:铜蓝蛋白10.4mg/dl,KF环(+),24小时尿铜168.9ug/24h,血清铜 0.29mg/L(0.7-1.4)。
基因检测结果:
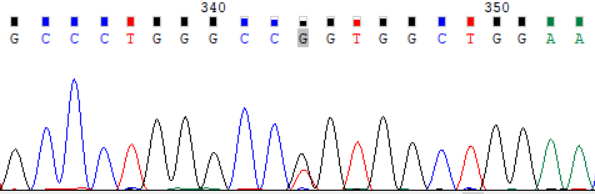
NCBI 参考序列:GCCCTGGGCCGGTGGCTGGAA
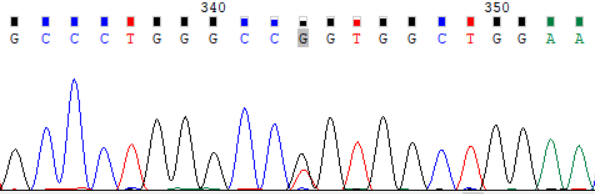
图 1:受检者 ATP7B_ex8 c.2333G>T (p.Arg778Leu) Sanger 测序验证图
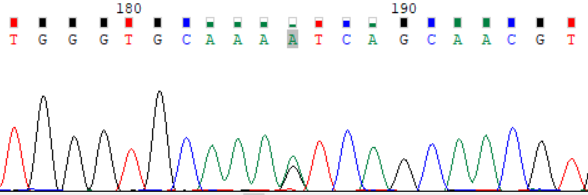
NCBI 参考序列:TGGGTGCAAAGTCAGCAACGT
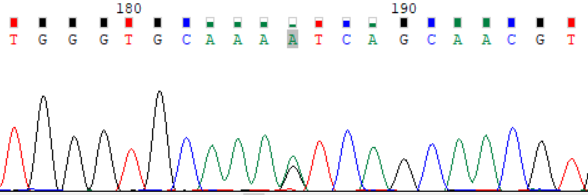
图 2:受检者 ATP7B_ex15 c.3316G>A (p.Val1106Ile) Sanger 测序验证图
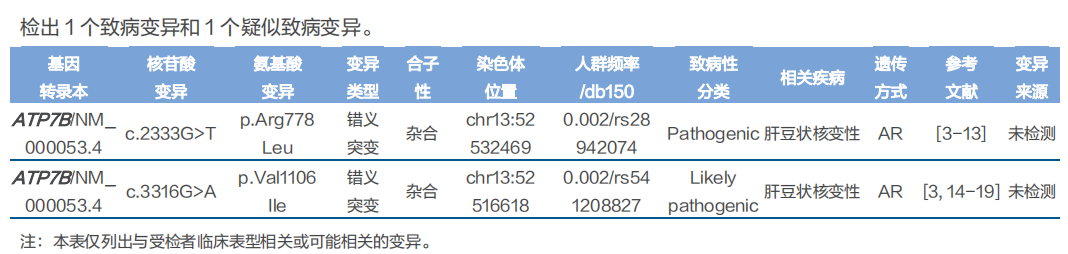
诊断:根据莱比锡评分,患者评分为10分,可以诊断肝豆状核变性。
治疗随访:青霉素皮试阴性后予以青霉胺(0.125g逐渐加量至0.75g每日)联合葡萄糖酸锌片(5片日三次空腹,),二者间隔至少2小时。同时辅以对症保肝、降酶、利尿药物治疗近2个月,白蛋白回升至35g/L,凝血活动度恢复至61%,腹水明显消退。复查24小时尿铜1623.7ug/24h。
白蛋白(10g/d) 病因治疗
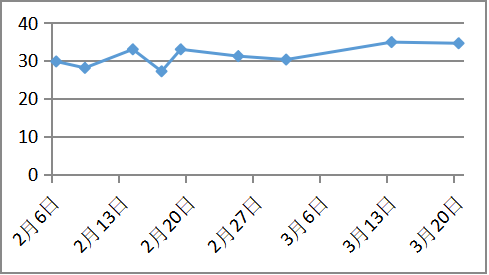
血浆白蛋白明显改善
病因治疗
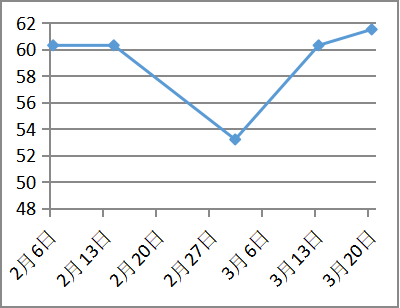
凝血功能改善
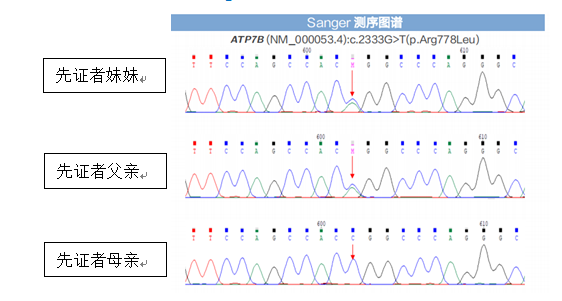
后续家系验证:先证者2个位点变异分别来自其父亲和母亲。先证者父亲、母亲肝功能、铜蓝蛋白均正常,未见KF环,无任何肝病症状。先证者妹妹经基因检测发现c.2333G>T(p.Arg778Leu)杂合变异,经验证来自其父亲,但其无肝病表现,肝功能示GGT31.4U/L,余指标正常,铜蓝蛋白、24小时尿铜正常。考虑先证者父母及妹妹为ATP7B基因变异携带者,有罹患肝豆状核变性的风险,建议定期检查评估是否出现肝豆状核变性相关症状,以便及时诊治。
先证者基因变异情况及来源
先证者父亲 ATP7B_ex8 c.2333G>T (p.Arg778Leu) Sanger 测序验证图
先证者母亲 ATP7B_ex15 c.3316G>A (p.Val1106Ile) Sanger 测序验证图
先证者妹妹基因变异情况及来源

先证者妹妹基因变异来源Sanger测序验证图
3.2 供稿专家简介

苏丽
承德医学院附属医院感染科主治医师
承德市感染性疾病质量管理与控制中心秘书
佑安专科联盟遗传代谢性肝病委员会委员
河北省肝病学会委员
擅长领域为常见感染性疾病及肝病的诊治
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间