
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:侯维
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
“国际罕见病日”自2008年起被设立于每年2月的最后一天,旨在提高人们对罕见病患者的认识,并为其提供支持帮助。2024年2月29日迎来了第17个国际罕见病日,中国罕见病联盟定义今年的主题为“关注罕见、点亮生命之光,弱有所扶、践行人民至上”。在这个特殊日子里,“遗传代谢性肝病月报”继续为您带来肝病临床一线的少见病例,特邀首都医科大学附属北京佑安医院孔明教授引领大家打开思路,跨学科思维认识黄疸。
一、主编致辞
遗传性血色病(hereditary hemochromatosis,HH)是一种全身性铁超负荷的潜在衰弱性遗传疾病,由参与调节铁稳态的基因突变引起。这些突变导致铁的吸收增加,导致铁在组织中过度沉积,最终导致器官损伤和疾病。HH导致的临床疾病谱,可能累及心脏(包括心肌病)、性腺功能减退、糖尿病、皮肤色素沉着、关节炎和肝纤维化[1]。
遗传性血色病根据致病基因的不同,可分为4个类型:1型也称HFE相关遗传性血色病,为染色体6p21.3上的HFE基因突变;2型分为2A及2B型,其中2A型致病基因为编码铁调素调节蛋白的HJV基因,2B型为编码铁调素的HAMP基因;3型致病基因为编码转铁蛋白受体2的TFR2基因;4型为编码膜铁转运蛋白(又称运铁素,Ferroportin)的SLC40A1基因。其中2~4型又称为非HFE相关血色病。1型血色病在具有欧洲血统的白种人中高发,患病率高达1/250-1/220[2]。我国原发性血色病总体少见,1型更为罕见,诊断的血色病多为非HFE相关血色病。
遗传性血色病2型也称为青少年型血色病(Juvenile hereditary hemochromatosis,JHH),是由HJV(2A型)或HAMP (2B型)基因突变引起的常染色体隐性遗传性疾病。HAMP基因编码铁调素,其功能缺失导致铁调素失活,铁调素负调控铁运素,仅占JHH病例的1/10[3]。关于HAMP基因相关突变,目前已被报道的主要突变位点包括93delG、c.148_150+1del、c.166C>T、c.175C>G、c.175C>T、c.176G>C、c.208T>C、c.223G>T、c.233G>A。
由HAMP突变引起的JHH通常会导致严重的铁过载和器官衰竭。文献中报道的病例通常发生在30岁之前,涉及多个系统:包括性腺功能障碍、心脏(包括心肌病)、腹痛、糖尿病、皮肤色素沉着、关节炎和肝纤维化,多数结局不佳。一旦诊断为JHH,应仔细评估和监测靶器官的潜在损伤,尽早保护重要器官清除铁沉积。静脉放血可以清除体内多余的铁, 周期性静脉放血在治疗铁超载方面比铁螯合剂更有效[4]。
本期月报报道1例HAMP突变的2B型遗传性血色病患者,此例患者发病较晚,发病年龄35岁,诊断年龄37岁,临床表现较轻,以转氨酶升高为主要表现,胃镜提示食管静脉曲张显露,血小板减少,考虑存在门脉高压。与大多数文献报道不同的是,本例患者并未出现关节、性腺、皮肤色素沉着、心功能异常及性腺激素异常的表现,其空腹血糖正常,仅糖耐量试验提示存在糖耐量减低的情况。本例患者经过放血治疗,病情得到很好控制,铁蛋白、血清铁和转铁蛋白饱和度均已恢复正常范围,肝功能持续正常。总之,本病例提示由HAMP突变引起的JHH可有多种临床表现,轻重不一,需要我们不断积累临床数据、探索疾病发病机制与基因型表达之间相关性。
参考文献:
1.Brissot P. Haemochromatosis[J]. Nat Rev Dis Primers, 2018, 4: 18016.
2.Wallace D F, Subramaniam V N. The global prevalence of HFE and non-HFE hemochromatosis estimated from analysis of next-generation sequencing data[J]. Genet Med, 2016, 18(6): 618-26.
3.Sandhu K. Phenotypic analysis of hemochromatosis subtypes reveals variations in severity of iron overload and clinical disease[J]. Blood, 2018, 132(1): 101-110.
4.Wu H X. Atypical juvenile hereditary hemochromatosis onset with positive pancreatic islet autoantibodies diabetes caused by novel mutations in HAMP and overall clinical management[J]. Mol Genet Genomic Med, 2020, 8(12): e1522.

侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.亚太肝脏研究协会:血色病的临床实践指南(Hepatology international, 2023,IF=6.60; Q1区)

2.基因筛查发现血色病后铁过载的监测和管理(JAMA network open, 2023, IF=13.80; Q1区)
3.Rusfertide治疗HFE相关血色病患者的铁过载:一项II期试验(Lancet Gastroenterol Hepatol, 2023, IF=35.70; Q1区)
4.儿童血色病:一个国家级回顾性队列(British journal of haematology, 2024, IF=6.50; Q1区)
5.美国遗传性血色病肝移植患者的长期预后和趋势(Liver transplantation, 2023, IF=4.60; Q1区)
6.HFE基因型、血色病诊断和80岁的临床结局:英国生物库的前瞻性队列研究(BMJ open, 2024, IF=2.90; Q2区)
7.综述:血色病(Lancet, 2023, IF=168.90; Q1区)
8.综述:血色病:铁死亡、ROS、肠道微生物和临床挑战(International journal of molecular sciences, 2024, IF=5.60; Q1区)
9.综述:主要的非HFE血色病(Journal of clinical and translational hepatology, 2023,IF=3.60; Q2区)
10.病例报道:HJV突变所致血色病:一对双胞胎的差异表型(Haematologica, 2024,IF=10.10; Q1区)
三、临床资讯
3.1 病例分享:1例轻型的HAMP基因突变致遗传性血色病
患者男性,37岁,因“发现转氨酶升高2年余”于2023年3月21日入我院肝病中心一科。
现病史:患者2年余前体检发现转氨酶升高,就诊于阜阳市医院复查肝功:ALT 101U/L ,AST 53U/L,腹部彩超提示:脾大。排除病毒性肝炎,给予“复方甘草酸苷片及肝喜乐”治疗。此后,患者长期服用多种保肝药物治疗,但期间多次复查肝功,转氨酶均未恢复正常。于半年前,为求明确诊治,就诊于外院门诊查: ALT 61U/L ,AST 79U/L,排除自身免疫性肝病、脂肪性肝病等,再次调整保肝药物“水飞蓟宾及双环醇”治疗三个月,后复查肝功仍未恢复正常(见图1)。
既往史:否认外伤、手术、输血史及食物药物过敏史。
个人史:无疫区疫水接触逗留史,无生鱼肉食用史,无吸烟史,无大量长期饮酒史。
家族史:其父母亲及弟弟均体健,子女体健。
查体:体温35.6℃,血压135/83mmHg,呼吸20次/分,身高4325px,体重53kg,体重指数17.7kg/m2。神志清楚,皮肤巩膜无黄染,心肺未见异常,腹软,肝脾未及,双下肢无水肿。
实验室检查:WBC 3.45×109/L,N% 0.62%,HGB 141g/L,PLT 77×109/L,网织红细胞绝对值 81×109/L;ALT 47U/L,AST 39U/L,TBIL 15.9μmol/L,DBIL 5.5μmol/L,γ-GT 38U/L,ALP 84U/L;PTA 62.4%,PT 11.2s。嗜肝病毒阴性,ANA 1:100,余均为阴性。免疫球蛋白均正常。甲状腺功能正常。AFP:2.27ng/ml(正常),CA199:113U/ml(正常值:≤30U/ml),PCT <0.05ng/ml(正常)。铜蓝蛋白:0.189g/L(正常值0.2-0.6g/L),铁蛋白:>2000 ng/ml(正常值30-400 ng/ml),转铁蛋白:1.43g/L(正常值2-3.6g/L),血清铁:43μmol/L(正常值10.6-36.7μmol/L),总铁结合力:62umol/L(正常),转铁蛋白饱和度:69.35%(正常值:男性<50%),糖耐量试验提示糖耐量减低(见图2)。性腺激素检测正常。
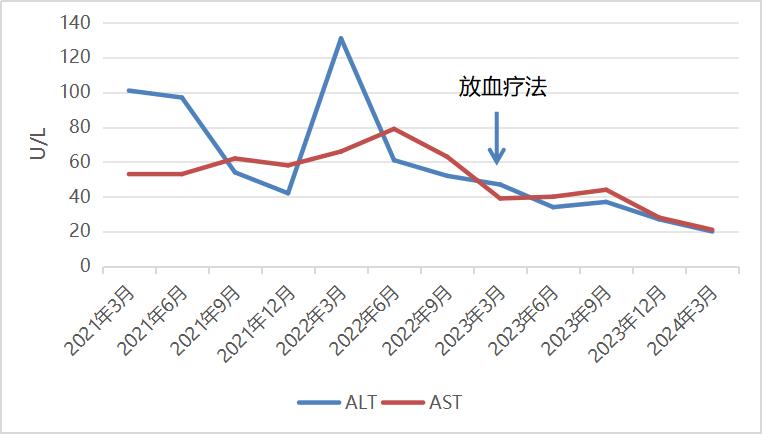
图1 患者转氨酶变化
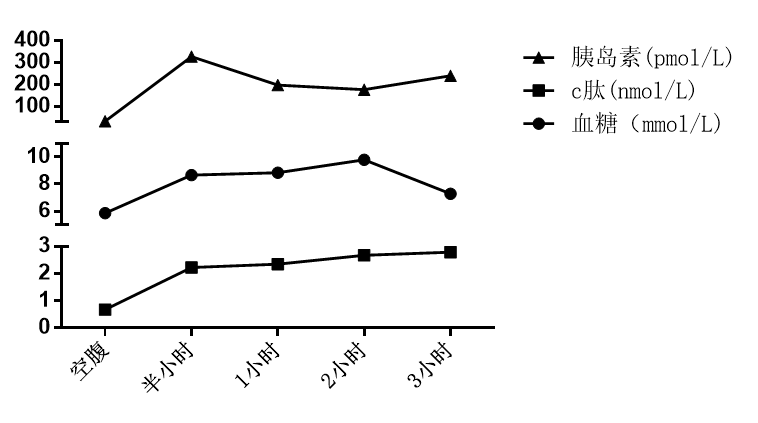
图2 患者糖耐量实验结果
影像学检查:心脏彩超及甲状腺彩超未见异常。B超提示肝脏回声增粗,脾大。Fibroscan:27kPa 。CT提示肝脏铁沉积(图3)。MRI提示肝脏、胰腺铁沉积表现,脾大(见图4)。胃镜示:食管静脉显露,慢性非萎缩性胃炎伴胆汁返流。
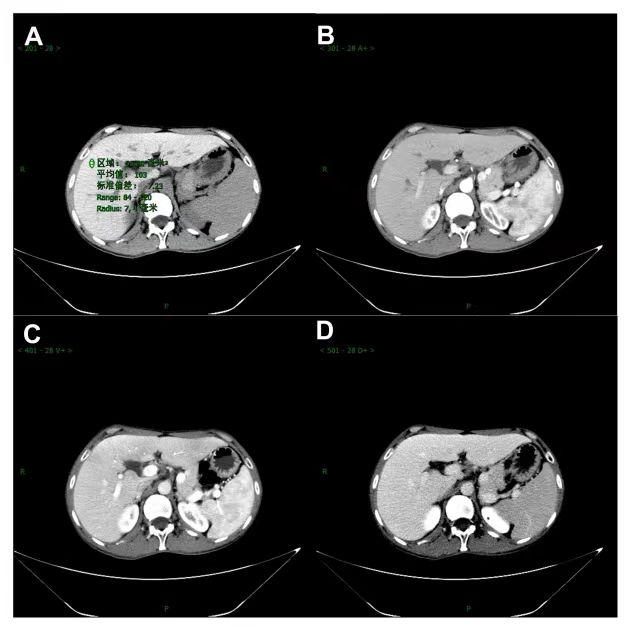
图3 腹部CT结果(A为平扫期,CT值为103;B为动脉期;C为门脉期;D为静脉期)
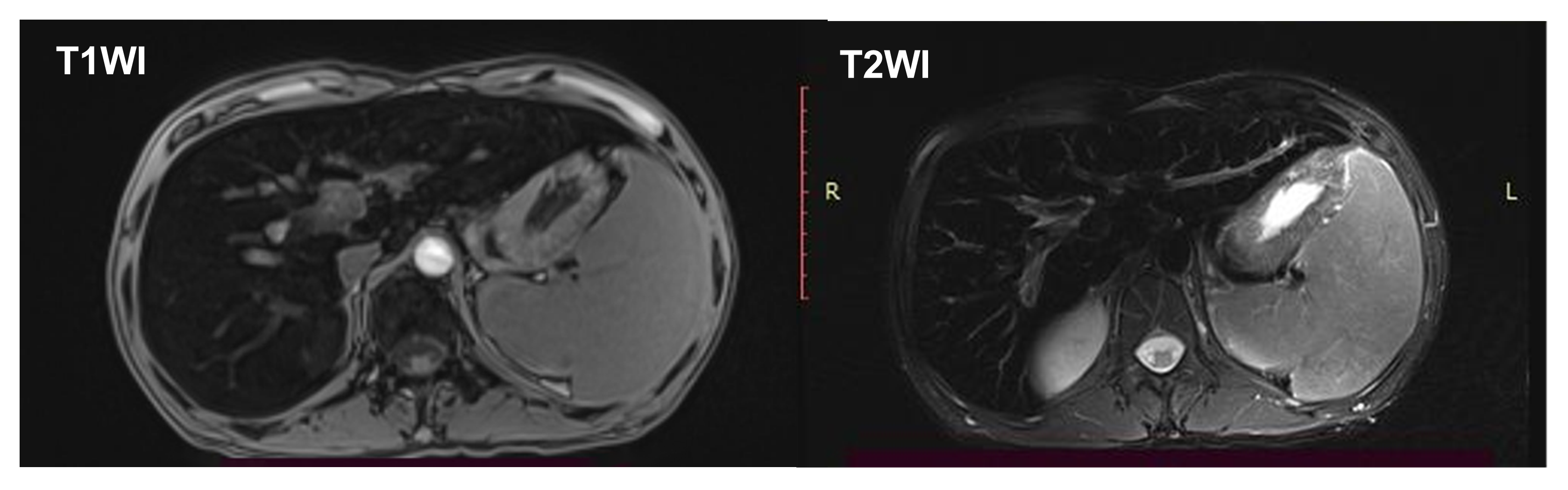
图4 腹部MRI结果T1及T2可见肝组织密度减低,提示肝脏铁沉积
肝穿病理:含铁血黄素沉积症,伴纤维化形成,纤维化程度相当于S3(见图5)。
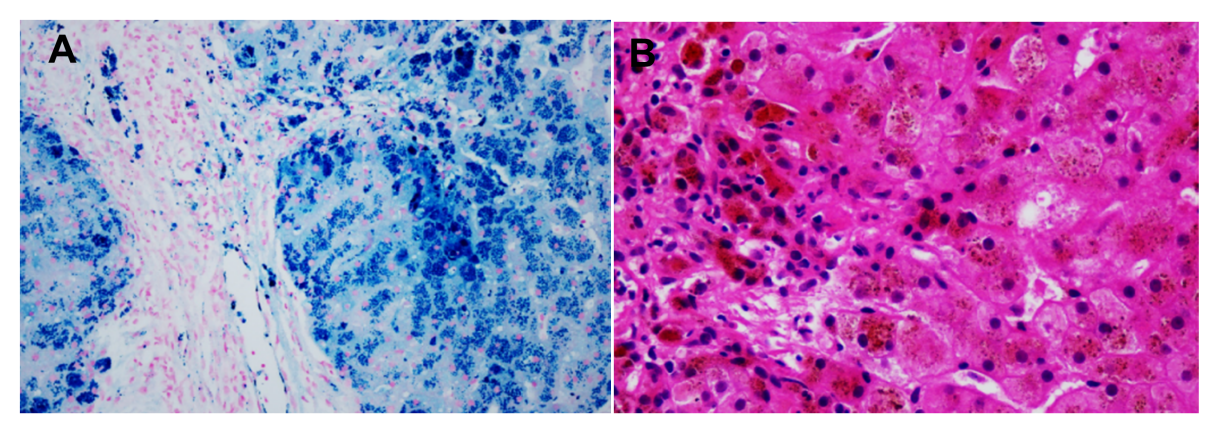
图5 肝活检显示含铁血黄素沉着并形成纤维化,纤维化程度相当于S3(A图普鲁士蓝染色,B图HE染色)
全外显子基因测序及家系验证:HAMP(NM_021175.4):c.166C>G(p.Arg56Gly)纯合突变(见图6),根据ACMG指南,PM2_Supporting+PP4+PM3_Strong,评估为可能致病。
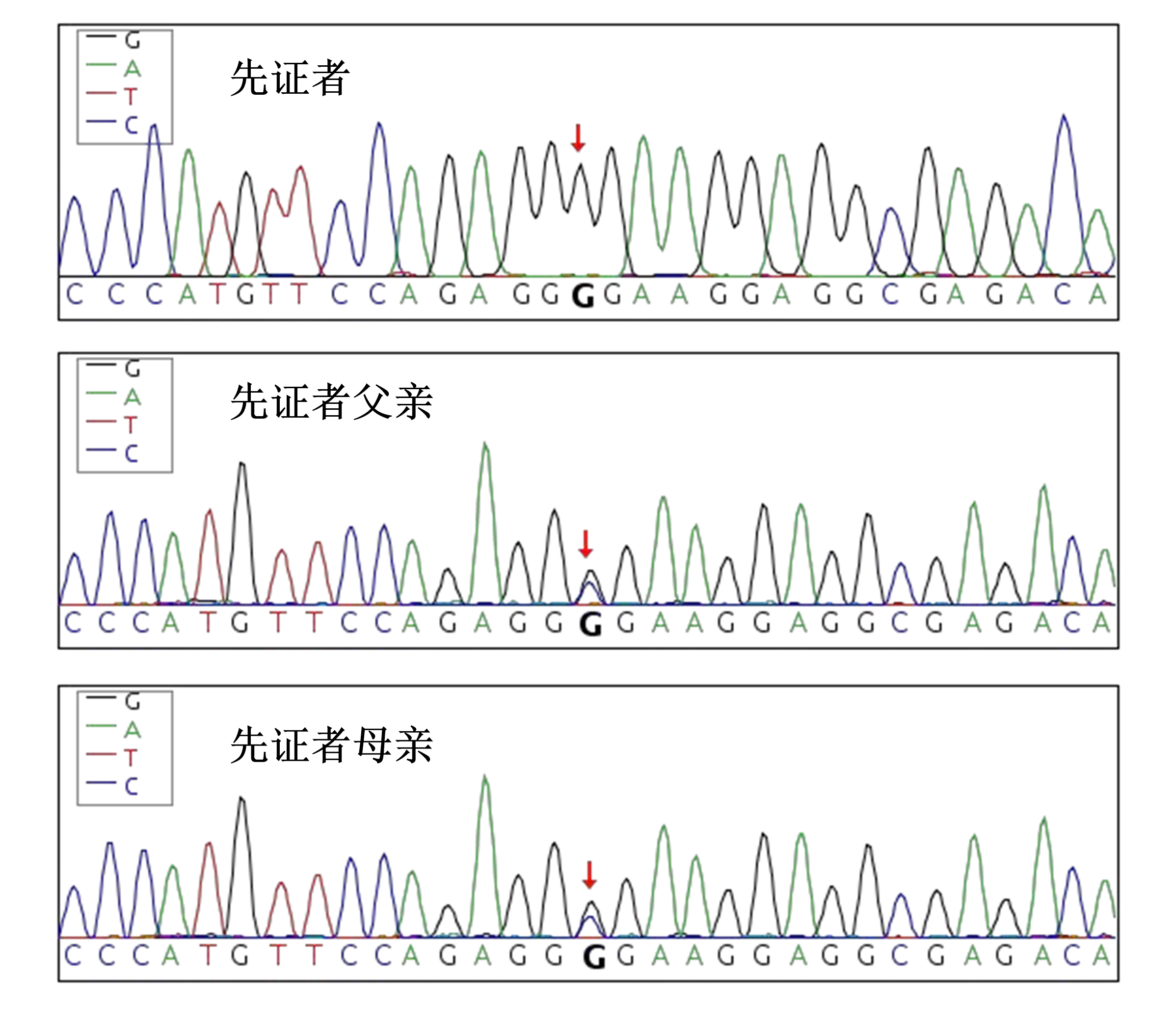
图6 基因检测提示患者HAMP:c.166C>G(p.Arg56Gly) 纯合突变,分别来自其父亲和母亲
最终诊断:结合患者肝功能异常,转铁蛋白饱和度升高,铁蛋白升高,腹部影像提示肝脏铁沉积,肝脏病理提示含铁血黄素沉着,基因检测存在HAMP基因c.166C>G(p.Arg56Gly)纯合突变,诊断“遗传性血色病(2B 型)”。
治疗与随访:给予患者静脉放血治疗,每周放血一次,每次350ml,并监测肝功能、血常规和铁蛋白。随访患者1年,目前肝功能已恢复正常,血红蛋白始终保持大于130 g/L,铁蛋白211.95μg/L(正常),血清铁18.1μmol/L(正常),总铁结合力54.3.4μmol/L(正常),转铁蛋白饱和度33.3%(正常)。空腹血糖在正常范围。
3.2 供稿专家简介
侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员

王征
首都医科大学附属北京佑安医院,肝病中心一科,副主任医师
主要研究方向为“遗传代谢性肝病”
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间