
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:韩英
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
糖原贮积症(glycogen storage disease,GSD)是由于糖原合成和分解代谢过程中各种酶缺陷导致的一组临床罕见的遗传代谢性疾病。其中GSD I型是最早发现且最常见的一型,为常染色体单基因隐性遗传病,发病率约为1/100000[1]。GSD I型可分为2种亚型:GSD Ia型致病基因G6PC位于染色体17q21,全长12.5 kb,包含5个外显子,编码葡萄糖-6-磷酸酶α(glucose-6-phosphatase-α,G6Pase-α);GSD Ib型致病基因SLC37A4,位于染色体11q23,全长5.3 kb,包含9个外显子,编码葡萄糖-6-磷酸转运体(glucose-6-phosphate transporter,G6PT)。大约80%的GSD I型患者为Ia型,20%为Ib型。
G6PT和G6Pase-α复合物可在肝、肾和肠细胞糖异生和糖原分解的最后一步催化细胞内葡萄糖-6-磷酸(glucose-6-phosphate,G6P)水解为葡萄糖。G6PT负责将G6P从细胞质运往内质网,G6Pase将G6P水解为葡萄糖和无机磷酸盐。GSD I型患者G6PC或SLC37A4基因突变均可使G6P向葡萄糖转变受阻,导致低血糖并促进糖原贮积,临床表现为肝肾肿大、腹泻等症状;由于糖代谢紊乱,可继发高乳酸血症,高脂血症,高尿酸血症等一系列的代谢异常。GSD Ib型患者还可以出现中性粒细胞减少、反复感染或炎症性肠病表现。GSD I型有两种较严重的远期并发症:肾功能不全和肝细胞腺瘤(hepatocellular adenoma,HCA),其中,有10%并发肝细胞腺瘤的患者可进展为肝细胞癌[2]。
诊断GSD I型应首先基于临床表现和生化异常,是否存在中性粒细胞减少可以帮助GSD Ia型和GSD Ib型诊断进行粗分类。既往GSD I型通过测量肝活检样本中G6Pase活性来确认。伴随基因检测技术发展,发现G6PC基因和SLC37A4基因2个等位基因致病突变具有确诊意义。
GSD I型诊断明确后应开始以饮食管理为主的治疗,治疗目标为维持血糖在正常范围。一般可采用餐间平均每4-6h补充生玉米淀粉,维持正常的餐间血糖,同时可以有效地缓解患儿的代谢异常。当饮食管理治疗不理想时,应辅以降脂药、降尿酸药缓解代谢异常,同时可使用ACE I类药物减轻肾小球高滤过状态,减缓肾功能恶化。对于GSD Ib患者,当中性粒细胞长期低于0.2×109/L时,可开始给予粒细胞集落刺激因子(G-CSF)治疗,但需注意长期G-CSF治疗会导致脾脏肿大,甚至出现骨髓异常增生综合征和急性髓系白血病。当发生肝细胞腺瘤并出现严重压迫、出血或有恶化迹象时,可行肝移植手术,若伴有肾功能衰竭,可同时行肝肾联合移植[1-2]。现阶段基因治疗仍处于探索阶段,用腺病毒载体介导G6PC或SLC37A4基因在动物体内进行治疗,并已进入临床试验阶段,初见疗效。
本期月报报道1例肝脏肿大原因待查的患儿,伴有生长迟缓和明显中性粒细胞减少,存在低血糖、高脂血症、高尿酸血症等代谢异常,经过基因检测最终诊断为糖原贮积症Ib型,我们还在该病例中发现了SLC37A4基因的一个新的剪接突变位点c.1123+1G>T。在诊治过程中有一个意外发现,该患儿在患GSD Ib的基础上合并银屑病,经过补充生玉米淀粉饮食治疗,不仅缓解了一系列代谢异常,而且银屑病病变几乎消失,GSD Ib与银屑病之间的关联有待进一步探索[3-5]。
参考文献:
1. Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat Rev Endocrinol 2010;6:676–88.
2. Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med 2014;16:e1.
3. Melis D, Carbone F, Minopoli G, et al. Cutting Edge: Increased Autoimmunity Risk in Glycogen Storage Disease Type 1b Is Associated with a Reduced Engagement of Glycolysis in T Cells and an Impaired Regulatory T Cell Function. J Immunol 2017;198:3803–3808.
4. Halprin KM, Okawara A, Levine V. Synthesis of glycogen in the psoriatic lesion. Arch Dermatol 1973;107:706–11.
5. Bai J, Zheng L, Han Y. Unexpected Findings in a Patient With Hepatomegaly and Skin Rashes. Gastroenterology 2018;155:e9–e12.

韩英
教授,博士生导师
空军军医大学西京医院消化内科主任
长江学者特聘教授
军队科技领军人才
中华医学会内科学分会前任主任委员
中华医学会肝病学分会副主任委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.中链甘油三酯和饮食干预改善了约旦1型糖原贮积症儿童的身体成分和代谢参数:一项临床试验(Foods, 2024,IF=5.20; Q1区)

2.恩格列净治疗糖原贮积症1b型患者的中性粒细胞减少症(Blood advances, 2024,IF=7.50; Q1区)
3.鉴定导致突尼斯糖原贮积症1b型患者的基因突变(Diabetology & metabolic syndrome, 2023,IF=4.80; Q2区)
4.关于IIIa 型糖原贮积症儿科患者的长期个性化高蛋白、高脂肪饮食(Journal of inherited metabolic disease, 2024,IF=4.20; Q1区)
5.接受酶替代疗法治疗的婴儿发病庞贝病患者的长期结果:一项德国-奥地利队列(Journal of neuromuscular diseases, 2024,IF=3.30; Q2区)
6.庞贝病新生儿患病率等筛查资料分析(Frontiers in pediatrics, 2024,IF=2.60; Q2区)
7.综述:评估 avalglucosidase alfa治疗迟发性庞贝病(Expert review of neurotherapeutics, 2024,IF=4.30; Q2区)
8.综述:戈谢病的女性患者(Biomedicines, 2024,IF=4.70; Q1区)
9.综述:基因治疗遗传性代谢性疾病(Journal of inherited metabolic disease, 2024,IF=4.20; Q1区)
10.病例报道:伴有中性粒细胞减少、马蹄肾和动静脉畸形的糖原贮积症 Ib 型患者中SLC37A4基因的新突变(Immunologic research, 2023,IF=4.40; Q2区)
三、临床资讯
3.1 病例分享:一例糖原贮积症Ib型患儿诊治中的意外发现
患儿男性,7岁,主因“肝脏肿大3年余”入西京医院消化内科。
现病史:患儿于3年余出现腹部膨隆,多食、易饿、易出汗,身高、体重较同龄儿童发育迟缓,曾就诊于当地医院行腹部超声检查提示肝大,间断服用“中草药”治疗效果不佳。2月前患儿无明显诱因体重下降约1.5kg,就诊于当地医院,查血常规示中性粒细胞绝对值 1.0×109/L(正常范围:1.8-6.3×109/L),生化示球蛋白41.6 g/L(正常范围:20-40g/L),余肝酶均正常,血糖3.34 mmol/L(正常范围:3.89-6.11mmol/L),甘油三酯 5.65 mmol/L(正常范围:0.28-1.8mmol/L),腹部超声和CT均提示肝脏肿大,骨穿示大致正常骨髓象,给予对症治疗后无明显缓解,门诊以“肝脏肿大待查”收入院。
既往史:2年前外院诊断“银屑病”,曾给予“中草药”治疗,效果不佳。外伤致上肢骨折2次,先后行手术治疗(具体不详)。
个人史: 生于陕西省,久居本地,无疫区疫水接触逗留史,无吸烟、饮酒史,未婚。
家族史:父亲、姑姑及祖母均患有“银屑病”,其父母四代以上有血缘关系,母及1哥1姐均体健,均无肝脏疾病史。
查体:身高110cm(同龄儿童第3百分位左右),体重20kg(同龄儿童第25百分位左右);头、面、颈部和躯干上可见散在红斑、皮屑;腹稍膨隆,腹围64.5cm,可见腹壁静脉显露,肝脏肋下12cm。
实验室检查:血常规:中性粒细胞绝对值 0.43×109/L(正常范围:1.8-6.3×109/L),余正常;肝功:球蛋白 40.1 g/L(正常范围:20-40g/L),余肝酶正常:ALT 18 IU/L(正常范围:9-50 IU/L),AST 22 IU/L(正常范围:15-40 IU/L),ALP 167 IU/L(正常范围:20-220 IU/L),GGT 25 IU/L(正常范围:10-60 IU/L);肾功:尿酸 706 μmol/L(正常范围:150-430 μmol/L),余正常;空腹血糖:3.10 mmol/L(正常范围:3.89-6.11 mmol/L);血脂:甘油三酯 8.08 mmol/L(正常范围:0.28-1.8 mmol/L),余正常;血沉:59 mm/h(正常范围:0-15mm/h);肌酸激酶32 IU/L(正常范围:50-310 IU/L);乳酸脱氢酶125 IU/L(正常范围:120-250 IU/L);尿酮体 1+;乙肝、丙肝阴性,EBV核及衣壳抗原IgG阳性,余病毒抗体均阴性;铜兰蛋白、24小时尿铜测定阴性;pANCA弱阳性,余自身抗体均阴性。
影像学检查:腹部超声示肝大,肝脏回声未见明显异常。
肝穿病理:肝小叶结构局部稍紊乱,肝细胞水肿,散在糖原化核肝细胞,局部肝细胞可见脂肪变性(微泡性为主),汇管区纤维组织轻度增生,极少量淋巴细胞、浆细胞浸润,小胆管结构存在(图1)。
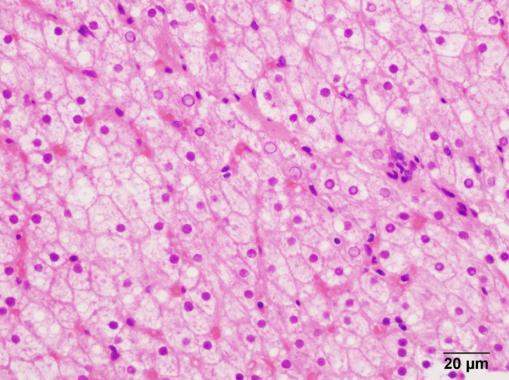
(图1.肝穿病理,HE染色)
全外显子基因测序及家系验证:根据上述临床特征,疑诊糖原贮积症I型,为进一步明确诊断并且进行疾病分型,完善基因测序。结果提示SLC37A4基因c.1243C>T(p.Arg415Ter)(已报道),杂合突变,ACMG评级为致病(PVS1+PM2_Supporting+PP4+PP5);SLC37A4基因c.1123+1G>T,杂合突变,ACMG评级为致病(PVS1+PM2_Supporting+PM3+PP4)。后经一代验证明确以上两种突变分别遗传自母亲和父亲(图2)。
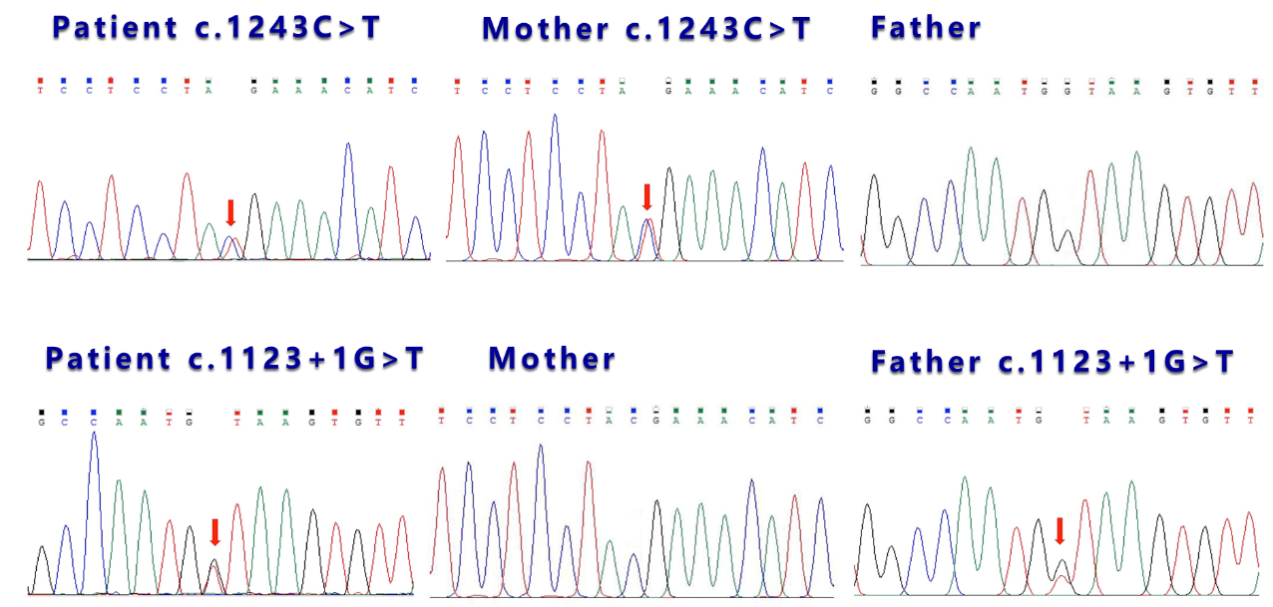
(图2.患儿及父母SLC37A4基因突变位点一代验证)
最终诊断:糖原贮积症Ib型。
治疗与随访:确诊后予患儿饮食疗法:以高碳水化合物食物为主,并在每餐之间加入生玉米淀粉(2g/kg,每天4次),以维持血糖正常范围。2月后复查显示患儿身高增长了9 cm,体重增加了3.5 kg。中性粒细胞绝对值轻度增加,空腹血糖增加,甘油三酯和尿酸显著降低。出乎意料的是,在没有任何针对银屑病的治疗下,银屑病皮损几乎消失(图3)。
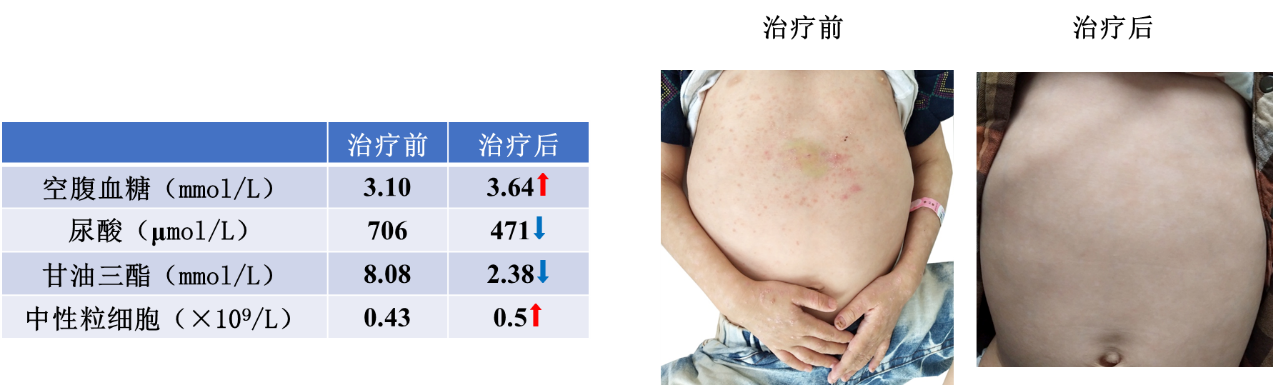
(图3.治疗与随访)
讨论:SLC37A4基因c.1123+1G>T突变位于外显子8和内含子8连接处的剪接位点(5’GU……AG-OH-3’),可能导致外显子跳跃或内含子保留,影响转录翻译过程。这可能是糖原贮积症Ib型的一种新突变,同时也证明了遗传异质性的存在。
该患儿在糖原贮积症Ib型的基础上合并银屑病,印证了糖原贮积症Ib型患者自身免疫性疾病患病风险增加的特征。在此病例中经过饮食疗法不仅缓解了一系列代谢异常,而且银屑病皮损几乎消失。既往文献表明银屑病病变中的糖原含量是正常表皮的5倍,结合本病例,考虑糖原贮积症Ib型与银屑病之间可能存在关联,但有待进一步探索。
(该病例报告Unexpected Findings in a Patient With Hepatomegaly and Skin Rashes已发表于Gastroenterology。)
3.2 供稿专家简介
韩英
教授,博士生导师
空军军医大学西京医院消化内科主任
长江学者特聘教授
军队科技领军人才
中华医学会内科学分会前任主任委员
中华医学会肝病学分会副主任委员

郑林华
空军军医大学西京医院 主治医师

白健
空军军医大学 临床遗传学博士 助理研究员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间