
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:侯维
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
遗传性血色病(hereditary hemochromatosis, HH)是一组以铁代谢异常而引起的全身性疾病,根据铁沉积部位及引起疾病程度不同,临床上可表现为肝硬化、糖尿病、皮肤色素沉着、充血性心力衰竭、性腺功能减退等多器官功能障碍。因肝脏在铁代谢中发挥重要作用,故为此类疾病的主要受累靶器官,若祛铁治疗不及时,约半数患者可死于肝细胞癌、消化道出血等严重肝病并发症。HH的诊断主要基于实验室、影像学及HH突变基因检测。早期发现并及时祛铁治疗,可延缓疾病发展,降低病死率。
HH根据其遗传基础主要可分为HFE基因突变型和非HFE基因突变型,根据致病突变基因具体分为以下4种类型[1-3]。1型在欧美人群中多见,是HFE基因突变所致的经典型血色病,为常染色体隐性遗传,其中C282Y基因变异最多见,其次为H63D基因变异,该型表现多样,90%患者无症状。2型为HJV基因和HAMP基因突变所致的血色病,为常染色体隐性遗传,前者编码铁调素调节蛋白,后者编码铁调素,此型患者发病率低,多见于近亲后代,多数起病早、病死率高。3型为编码转铁蛋白受体2的TFR2基因突变所致的血色病,为常染色体隐性遗传。4型为SLC40A1基因突变所致的血色病,该基因编码膜铁转运蛋白(ferroportin,FPN),与前3型的遗传特征不同,4型血色病为常染色体显性遗传[4]。FPN通过控制肠细胞和巨噬细胞来介导铁的跨膜输出过程,若表达降低会致细胞内游离铁增加,对机体造成影响。Pietrangelo[5]在2017年建议将SLC40A1突变所致的HH分为两类,即:功能缺失型(4A型)和功能获得型(4B型),前者使FPN不能定位在细胞表面,使铁由胞内向胞外转出受阻,进而沉积在巨噬细胞中,间质细胞铁过载,临床上常表现为血清铁降低、转铁蛋白饱和度正常或降低,MRI易出现“黑脾” ,该类型放血治疗疗效不佳,且易并发贫血;后者FPN可以定位于细胞表面,但不能与铁调素相结合,引起铁调素抵抗,造成FPN蓄积,将过多的铁从细胞内转运入血浆,并在肝脏实质内沉积。
本期月报报道1例遗传性血色病4A型患者,该患者起病隐匿,仅表现为血小板减少,转铁蛋白饱和度基本正常,腹部核磁提示肝脏、脾脏铁沉积,基因检测证实SLC40A1基因致病性杂合变异。予患者完善检查评估其他脏器铁过载受累情况,其中性腺及心脏功能未见明显异常,口服葡萄糖耐量试验提示糖耐量减低,但是否与铁代谢异常相关目前证据不足。本例患者经过地拉罗司治疗2年后铁蛋白无明显下降,后尝试放血治疗,期间监测血红蛋白保持正常范围,患者对放血治疗耐受性良好,虽然半年后随访铁蛋白从9668ng/ml降至7466ng/ml,但放血总疗程尚短,需进一步动态观察评估效果。
我们还对本病例进行了家系筛查,并发现家系中多位成员均携带此致病基因。有研究认为,HH的临床病程大致分为三个阶段:第一阶段指存在基因及其他易感因素,但尚无铁指标异常;第二阶段出现铁指标异常,但尚未出现组织学损伤的表现;第三阶段铁过载已导致组织或器官出现损伤或并发症[6],此阶段常出现明显的临床表现,祛铁治疗能改善铁超载状态,对于已造成的组织损伤不能完全逆转。本病例于体检时发现血小板水平低下,进一步检查确诊4A型HH,家族中其他致病基因携带者均出现铁指标异常,部分出现组织学损伤表现,并已开始治疗。
参考文献:
[1]Zoller H., Schaerer B., Vanclooster A., e tal. EASL Clinical Practice Guidelines on haemochromatosis. J Hepatol, 2022,77(2):479-502.
[2]中国2型糖尿病防治指南(2020年版)[J].中国实用内科杂志,2021,41(09):757-784.
[3]韩悦,张欣欣.遗传性血色病的基因诊断[J].临床肝胆病杂志,2019,35(08):1673-1679.
[4]Pietrangelo A. Hereditary hemochromatosis: ptheogenesis, dignosis, and treatment. Gastroenterology, 2010,139(2):393-408.
[5]Pietrangelo A. Ferroportin disease: Pathogenesis, diagnosis and treatment[J]. Haematologica, 2017,102(12):1972-1984.
[6]Adams P, Brissot P, Powell LW, EASL International Consensus Conference on haemochromatosis, J Hepatol,2000,33(3):485-504.

侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.铁、血色病基因型和感染风险:一项针对 142 188 名普通人群个体的队列研究(Blood, 2024, IF=21.0; Q1区)

2.HFE型血色病的外显率、癌症发病率和生存率(Liver international, 2024, IF=6.00; Q1区)
3.SUGP2 p.(Arg639Gln)变异通过 CIRBPBMPER 信号通路参与血色病的发病机制(American journal of hematology, 2024, IF=10.10; Q1区)
4.FPN1 铁输出因子的功能双重丧失和获得导致铁转运蛋白病表型(HGG advances, 2024, IF=3.30; Q2区)
5.是否应该进行遗传性血色病的人群筛查(Gene, 2024, IF=2.80; Q2区)
6.综述:血色病 - 如何不忽视并妥善管理“铁人”(Journal of clinical medicine, 2024, IF=3.00; Q1区)
7.综述:迟发性皮肤卟啉病的临床管理的最新进展(Liver international, 2024, IF=6.00; Q1区)
8.系统评价:血色病筛查和治疗的健康经济情况(PharmacoEconomics, 2024, IF=2.20; Q2区)
9.病例报道:一例遗传性血色病 3 型的日本患者检出转铁蛋白受体 2 的新突变(Hepatology research, 2024, IF=3.90; Q1区)
10.病例报道:一例70岁无症状的铁转运蛋白病女性患者(Internal medicine, 2024, IF=1.00; Q3区)
三、临床资讯
3.1 病例分享:1例遗传性血色病4A型患者
患者男性,63岁,主因“发现血小板减少2年余”于2024-3入首都医科大学附属北京佑安医院住院。
现病史:患者2年余前体检时发现血小板减少,当地医院血液科就诊,骨髓活检提示:骨髓组织增生活跃,红系比例增高,易见含铁血黄素沉积,巨核细胞多见,基因检查显示SLC40A1 NM_014585:c.364A>T(p.Arg88Ser)杂合突变。化验血清铁156µmol/L(参考范围:10.6-36.7 µmol/L),铁蛋白2021ng/ml(参考范围:30-400ng/ml),CT提示脾脏密度增高,开始口服地拉罗司治疗,监测铁蛋白下降不理想,现为进一步治疗就诊我院。自发病以来体重下降约3kg。
既往史、个人史及家族史:高血压8年,规律服用缬沙坦80mg/d,瑞舒伐他汀10mg/d。吸烟50余年,5-7支/天,戒烟1月,偶社交性吸烟及饮酒。否认疫区、疫水接触史,否认手术、输血、冶游史,否认肝病家族史。
入院查体:T 36.3℃,P 83次/分,R 20次/分,BP 95/79mmHg。神志清楚,面色晦暗,皮肤巩膜未见黄染,上腹部触诊稍韧,腹部无压痛及反跳痛,肝脏及脾脏未触及,移动性浊音(-),肝区叩击痛(-),墨菲氏征(-),双下肢无水肿。余查体未及异常。
入院后辅助检查:
血常规:WBC 4.67×109/L,RBC 3.76×1012/L,HB 131g/L,PLT 30×109/L;
尿常规:PRO(-)、BLD(-)、PH 6.0;
血生化:ALT 44U/L、AST 42U/L、ALB 33.9g/L、CHE 4685U/L、TBil 9.5µmol/L、DBil 4.4µmol/L、Cr 50µmol/L、Glu 4.52mmol/L、Ca 2.15mmol/L;
凝血功能:PTA 86.7%,D-Dimer< 0.2ug/L;
病原学检查:HBsAb(+)、HBcAb(+),HIV(-)、梅毒(-)、HCV(-)、HEV IgG(+)、HEV IgM(-)、CMV IgG(-)、CMV IgM(-)、HEV PCR(-)、HAV IgM(-)、CMV DNA(-)、EBV DNA(-);
免疫学相关检查:IgG 14.2/L、IgA1.83g/L、IgM0.801g/L(均在正常范围);ANA1:100(胞浆颗粒),余自身抗体均为阴性;
其他肝脏相关检查:铜蓝蛋白0.229g/L、甲胎蛋白1.34ngml、血氨51µg/dl(均在正常范围);
铁代谢指标:血清铁28µmol/L(参考范围:10.6-36.7 µmol/L)、铁蛋白>2000ng/ml(参考范围:30-400ng/ml)、总铁结合力55µmol/L(正常范围)、未饱和铁结合力31µmol/L(正常范围)、转铁蛋白饱和度50.9%(参考范围20-50%);
其他脏器功能化验:
心脏:CK 28U/L、CK-MB 1.53ng/ml、TNI 0.007ng/ml、LDH 148U/L、BNP 31pg/ml(均在正常范围);
性腺:性激素测定未见异常;
甲状腺及甲状旁腺:甲状腺及甲状旁腺激素测定未见异常;
胰岛:HbA1c 5.0%(正常范围);口服葡萄糖耐量实验(OGTT):提示糖耐量减低,见表1。
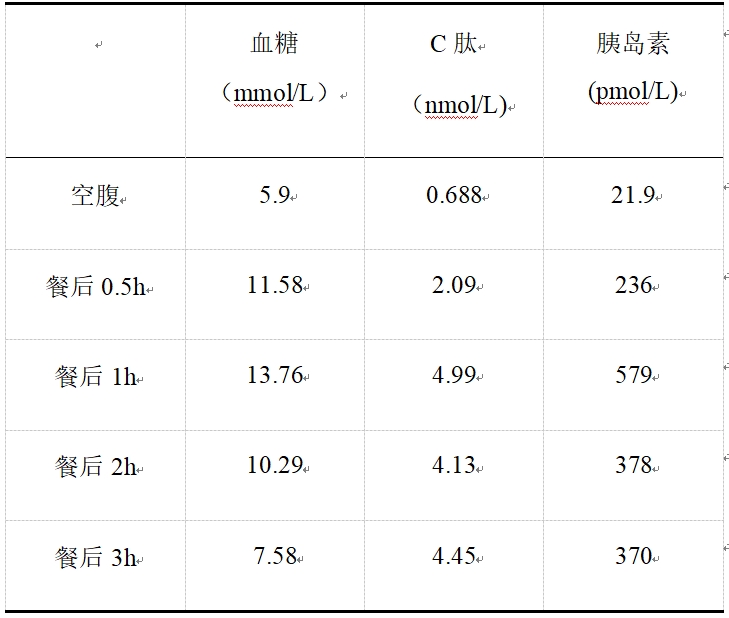
表1 患者OGTT结果
影像学检查:
胸部CT平扫:双肺多发小结节,动脉硬化。
腹部B超:弥漫性肝病表现,脾大,脾静脉增宽(门静脉内径12mm、脾厚46mm、脾静脉内径10mm)。
肝脏弹性测定:E10.4kPa,CAP 245 dB/m。
胃镜:食管上中下段黏膜光滑,呈粉红色,未见糜烂、溃疡及静脉曲张,慢性萎缩性胃炎(C2)。
心脏彩超:三尖瓣返流(少量),左室舒张功能减低,LVEF60%。
腹部增强MRI:肝脾铁过载、右肾小囊肿。(见图1)
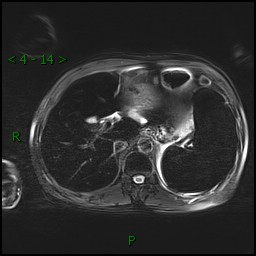
图1 腹部增强MRI(注:T2W1肝信号减少,与脾脏大致相同,在MRI扫描上表现为“黑肝+黑脾”)
颅脑MRI:右侧侧脑室前角条状影,左侧乳突炎可能。
肝穿病理:肝穿组织1条,小叶结构紊乱,纤维组织增生并分隔肝实质,局灶呈结节状;切片内见12个中小汇管区及6个较大汇管区,间质中度混合性炎细胞浸润,以淋巴细胞为主,可见散在浆细胞、嗜酸性细胞浸润,其中1个汇管区淋巴细胞密集伴较多浆细胞浸润,可见轻度界面炎;部分汇管区小胆管肿胀、部分蔽塞,周围见细胆管反应性增生,Masson及网织染色示汇管区胶原纤维增生;D-Pas染色示汇管区及肝小叶内见较多蜡质样细胞;普鲁士蓝染色显示汇管区及肝腺泡I-III带库普弗细胞及肝细胞内见弥漫性铁颗粒沉着,以库普弗细胞沉积为主,累积全部肝小叶,肝实质可见点灶状坏死,局部肝窦扩张。诊断:肝内铁过载,4级,考虑遗传性血色病(IV型)并肝纤维化S3。特殊染色:普鲁士蓝(铁+);罗丹宁(铜-)(见图2-3)。
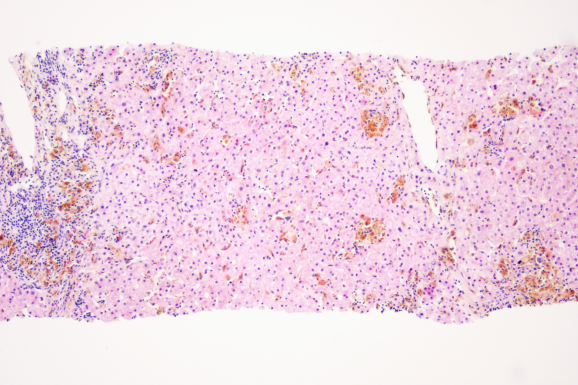
图2 HE染色 10×
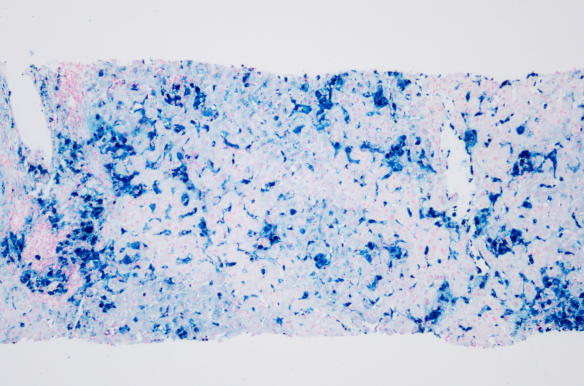
图3 普鲁士蓝染色 10
全外显子基因检测(2022年5月):SLC40A1基因NM_014585:c.364A>T(p.Arg88Ser),杂合突变,根据ACMG指南评级为可能致病(PM1+PM2+PM5+PP1+PP3+PP4)。
家系调查:对患者进行家系调查,患者父亲因肺心病于60岁时死亡,是否患有血色病无法追溯,母亲正常。先证者一子带有同样变异基因,已发病(表现为肝功异常,铁蛋白7410ng/ml,放血治疗一年半,每2周一次,400ml/次,目前铁蛋白降至4600ng/ml,HB 133g/L);先证者4个兄弟姐妹中有一妹妹带有同样变异基因,且已发病(ALT 48 AST 43 TBIL 8.2 ALB 33.6 铁蛋白>2000ng/ml,血清铁 28μmol/L,肝弹性10.4kPa,放血治疗4个月,不能耐受,出现头晕、乏力等表现,HB128g/L,目前口服地拉罗司治疗),此妹妹的3个子女中有一子、一女带有此变异基因,出现转氨酶升高,具体情况不详(图4)。
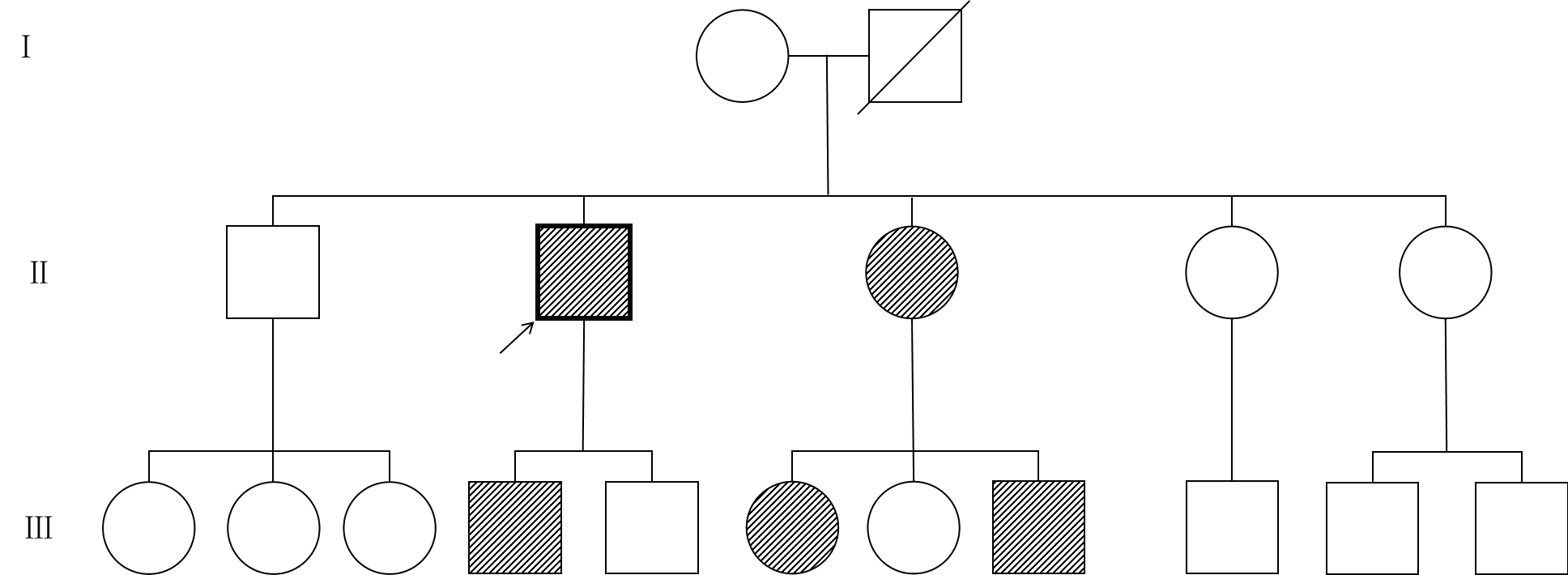
图4 患者家系图(箭头所指为先证者)
诊断:患者中年男性,起病隐匿,病程中肝功能正常,仅出现血小板减少。化验铁蛋白升高,此次住院血清铁正常,转铁蛋白饱和度基本正常。腹部核磁可见肝脏及脾脏均为低信号(黑肝+黑脾)。肝穿病理示汇管区及肝腺泡I-III带库普弗细胞及肝细胞内弥漫性铁颗粒沉着,以库普弗细胞沉积为主,肝纤维化S3。基因检测提示SLC40A1基因存在致病性杂合变异。结合以上表现,除外肿瘤、急慢性炎症、血液系统疾病等铁过载的继发因素,考虑诊断遗传性血色病4A型。
治疗及随访:根据指南推荐意见及4A型血色病疾病特点,放血治疗不作为4A型血色病首选的祛铁治疗方案,故本病例发病初期应用铁螯合剂治疗,但因存在明显的消化道症状,长期不能耐受药物,且治疗2年期间铁蛋白下降不理想,患者于2024年3月11日首次接受放血200ml治疗,并停用地拉罗司。随访至2024年9月,患者总计放血8次,每次间隔2-4周,放血前均进行血常规检测,血红蛋白均大于120g/L,治疗期间无不适,患者对放血治疗耐受性良好,虽然半年后随访铁蛋白从9668ng/ml降至7466ng/ml,但放血总疗程尚短,需进一步动态观察评估效果。
3.2 供稿专家简介
侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员

王克菲
首都医科大学附属北京佑安医院,肝病中心一科,主治医师
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间