
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张岭漪
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
先天性肝纤维化(Congenital Hepatic Fibrosis,CHF)在1961年由Kerr教授报道并命名,被认为是一种罕见的常染色体隐性遗传性疾病,该病与胆管板畸形(Ductal Plate Malformation, DPM)所致的肝内胆管遗传发育障碍有关[1]。根据不同的组织分型可表现出不同的临床症状,诊断需要结合肝组织病理和基因检测。
CHF是一种主要影响肝脏和肾脏的纤维囊性疾病,其特点是胆管板畸形以及肾集合管呈梭形扩张,发病率约为1:20000。CHF作为一种罕见的常染色体隐性遗传疾病,大多数病例是由PKHD1基因突变引起[2-4]。目前已报道该基因突变位点及组合已超过800余种,该基因编码纤维囊肿蛋白/多管蛋白(Fibrocystin/Polyductin, FPC),当PKHD1基因发生突变后便会引起胆管细胞FPC蛋白功能缺陷,从而导致胆管板发育畸形[5]。近期亦有学者提出CHF是由PKHD1基因突变、胆管板细胞极性改变以及β-catenin依赖的趋化因子分泌增加引起的一种遗传性胆管病变,并且发现在PKHD1基因突变小鼠CHF模型中,Scribble/YAP/β-catenin轴的失调维持了纤维炎症反应[6-8]。胆管板是胆道系统的起源,其缺陷导致胚胎胆管过多和门静脉分支异常。在典型的CHF中,胆管板缺损位于直径较小的小叶间胆管水平,而直径较大的肝内胆管发育异常则在Caroli’s病患者中更多见。许多CHF患者有一种或多种相关的胆道异常,包括胆管错构瘤、Caroli’s病以及肝外胆管梭状扩张,因此易引起复发性胆管炎。此外,本病亦可引起血管异常包括肝动脉肿大、门静脉海绵样变性以及门静脉血栓形成,此为引起非肝硬化性门静脉高压的原因[9]。纤维囊性肾病和髓质海绵肾为代表的肾脏异常,也是CHF常伴随的临床特征之一[10]。
临床上将CHF分为4种类型,分别是门静脉高压型、胆管炎型、门静脉高压与胆管炎混合型和隐匿型[11]。其中门静脉高压型和混合型较多见,而胆管炎型和隐匿型相对罕见。当出现重叠的异常胆管囊性改变时,则称为Caroli综合征[12]。
目前,有个案报道提到通过经颈静脉肝内门腔静脉分流术治疗门静脉高压型CHF患者,能在一定程度上降低消化道出血的风险[13-14]。但对于胆管炎型CHF患者,目前肝移植仍然是最佳的根治方案。
本期月报报道一例青年男性CHF患者,既往曾多次发作胆管炎,肝脏生化指标基本正常,影像表现为胆管走行异常,更重要的是病理学检查发现汇管区间质大量纤维组织增生,伴明显胆管板畸形,考虑CHF。基因检测证实PKHD1基因c.3071T>G纯合变异。虽然此位点既往未见报道,ACMG评分为uncertain,但该患者符合胆管炎型CHF的临床表现,将来可进一步进行功能验证来明确。临床上对于不明原因的门静脉高压患者,应考虑到CHF的可能,而对于反复发作胆管炎的年轻患者,我们亦不能排外CHF,更不能简单的仅通过影像学诊断为胆囊炎或伴胆管炎。希望通过分享本例患者的诊治过程,能增强肝胆科医师对CHF疾病认识,提高该病的早期诊断率。
参考文献:
[1] Kerr DN, Harrison CV, Sherlock S, et al. Congenital hepatic fibrosis[J]. Q J Med, 1961, 30(117):91-117.
[2] Rawla P, Sunkara T, Muralidharan P, et al. An updated review of cystic hepatic lesions[J]. Clin Exp Hepatol, 2019, 5(1):22-29. DOI:10.5114/ceh.2019.83153.[3] Olaizola P, Rodrigues PM, Caballero-Camino FJ, et al. Genetics, pathobiology and therapeutic opportunities of polycystic liver disease. Nat Rev GastroenterolHepatol,2022,19(9):585-604. DOI:10.1038/s41575-022-00617-7.
[4] Mirza H, Besse W, Somlo S, et al. An update on ductal plate malformations and fibropolycystic diseases of the liver[J]. Hum Pathol, 2023, 132:102-113. DOI:10.1016/j.humpath.2022.06.022.
[5] Raynaud P, Tate J, Callens C, et al. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis[J]. Hepatology, 2011, 53(6):1959-1966. DOI:10.1002/hep.24292.
[6] Duspara K, Bojanic K, Pejic JI, et al. Targeting the Wnt signaling pathway in liver fibrosis for drug options: an update[J]. J Clin Transl Hepatol, 2021, 9(6):960-971. DOI:10.14218/jcth.2021.00065.
[7] Fabris L, Milani C, Fiorotto R, et al. Dysregulation of the scribble/YAP/β-catenin axis sustains the fibroinflammatory response in a PKHD1(-/-) mouse model of congenital hepatic fibrosis[J]. Faseb J, 2022, 36(6):e22364. DOI:10.1096/fj.202101924R.
[8] Drögemüller M, Jagannathan V, Welle MM, et al. Congenital hepatic fibrosis in the Franches-Montagnes horse is associated with the polycystic kidney and hepatic disease 1 (PKHD1) gene[J]. PLoS One, 2014, 9(10):e110125. DOI:10.1371/journal.pone.0110125.
[9] Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: a review of the literature[J]. World J Gastroenterol, 2010, 16(6):683-690. DOI:10.3748/wjg.v16.i6.683.
[10] 杜白雪, 李东颖, 马怡辉, 等. 先天性肝纤维化并髓质海绵肾1例报道[J]. 胃肠病学和肝病学杂志, 2016, 25(12):1384-1385. DOI: 10.3969/j.issn.1006-5709.2016.12.020.
[11] Lasagni A, Cadamuro M, Morana G, et al. Fibrocystic liver disease: novel concepts and translational perspectives[J]. Transl Gastroenterol Hepatol, 2021, 6:26. DOI:10.21037/tgh-2020-04.
[12] Acevedo E, Laínez SS, Cáceres Cano PA, et al. Caroli's syndrome: an early presentation[J]. Cureus, 2020, 12(10):e11029. DOI:10.7759/cureus.11029.
[13] Yang Y, He C, Yuan X, et al. Portal fibrotic cord is associated with transjugular intrahepatic portosystemic shunt failure and death in cirrhotic patients[J]. J Clin Transl Hepatol, 2023, 11(4):809-816. DOI:10.14218/jcth.2022.00391.
[14] Van Hoek B. Defining the value of extracorporeal liver support in acute and acute-on-chronic liver failure[J]. J Clin Transl Hepatol, 2023, 11(3):517-520. DOI:10.14218/jcth.2022.00025.

张岭漪
主任医师,教授,研究生导师
兰州大学第二医院肝病科 主任
中华医学会肝病学分会、预防感染病防控分会委员
中华医学会肝病学分会肝纤维化学组委员、终末肝学组委员
中国医疗保健国际交流促进会、肝胆疾病学分会常务委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.成年期诊断的先天性肝纤维化的临床评价(Annals of hepatology, 2024, IF=3.70; Q2区)

2.肝脏胆管板畸形和纤维囊性疾病的最新进展(Human pathology, 2023, IF=2.70; Q2区)
3.2832个JAG1变异的功能表征支持Alagille 综合征的重新分类,并改善了临床变异解读的指导(American journal of human genetics, 2024, IF=8.10; Q1区)
4.非常罕见的NOTCH2变异与 Alagille 综合征临床特征的关联(Gene, 2024, IF=2.80; Q2区)
5.综述:遗传性慢性胆汁淤积性肝病(Journal of clinical and translational hepatology, 2024, IF=3.10; Q2区)
6.病例报道:一例罕见的模拟先天性肝纤维化病例(Gastroenterology, 2024, IF=25.70; Q1区)
7.病例报道:一例非典型的 Caroli 综合征诊断(Diagnostic pathology, 2024, IF=2.40; Q2区)
8.病例报道:一例WDR19基因突变致肝内胆管扩张表现为Caroli综合征(Translational pediatrics, 2024, IF=1.50; Q2区)
9.病例报告:一例成人常染色体显性遗传性多囊肾合并先天性肝纤维化(Frontiers in medicine, 2024, IF=3.10; Q1区)
10.病例报道:Alagille 综合征的挑战和见解(Gastroenterology report, 2024, IF=3.80; Q2区)
三、临床资讯
3.1 病例分享:先天性肝纤维化1例
患者,男性,27岁,主诉“乏力、发热10天”。
现病史:患者于入院前10天无明显诱因出现乏力、发热,伴有间断口干、寒战,体温最高达38.3℃,物理降温可恢复正常,但仍伴有间断上腹部疼痛,并放射至右肩背部。急诊检查:血常规示:白细胞计数7.4x109/L,中性粒细胞比率0.86,血红蛋白92g/L,血小板计数423 x109/L;炎症指标:C反应蛋白291.82mg/L,降钙素原2.68ng/ml,血沉:118mm/h,白细胞介6 343pg/ml;生化指标:白蛋白36.6g/L,总胆红素33.7μmol/L,直接胆红素26.2μmol/L,γ-谷氨酰转移酶129U/L,碱性磷酸酶367U/L,转氨酶正常;尿常规:尿蛋白(++),尿胆红素(+),尿胆原(+++)。腹部彩色多普勒超声提示:肝大,脾大,肝内多发高回声病灶,血管瘤可能;胆囊体积小;胰、双肾未见明显异常。为进一步诊治收入我科。
既往史:有反复胆管炎发作史。
入院查体:体温39. 3℃ ,脉搏 106 次/min,呼吸 20次/min,血压 118/62 mmHg。轻度贫血貌,全身皮肤黏膜无黄染。浅表淋巴结未触及肿大,无肝掌、蜘蛛痣。心、肺查体无特殊。腹软,右上腹部轻压痛,无反跳痛,全腹未触及包块,肝脾肋下未触及,Murphy’s征阳性,移动性浊音阴性,肠鸣音正常,双下肢无水肿。
入院后化验与检查:
感染相关化验:甲、乙、丙、戊型肝炎病毒标志物、HBV DNA、EBV病毒、弓形体抗体、风疹病毒、巨细胞病毒、单纯疱疹病毒1型及2型、结核杆菌感染T细胞检测试验、布鲁氏菌试验均阴性;血培养未检出阳性细菌;宏基因检测:肺炎克雷伯杆菌。
其他化验:INR 1.55;肝病自身抗体+免疫球蛋白、铜蓝蛋白、甲胎蛋白检查均正常。
骨髓穿刺检查:骨髓三系(巨核细胞、红系细胞和粒细胞)增生。
腹部MRI+胰胆管磁共振成像检查:肝内胆管走行僵硬、管壁毛糙并不均匀迂曲扩张(尤其肝右叶胆管),胆管炎多考虑,硬化性胆管炎?慢性胆囊炎,胆总管下端结节并胆道轻度梗阻性扩张,炎性改变多考虑;同时可见脾脏轻度肿大,门静脉宽度正常,未见门静脉高压征象(图1)。
胃镜:未见异常。
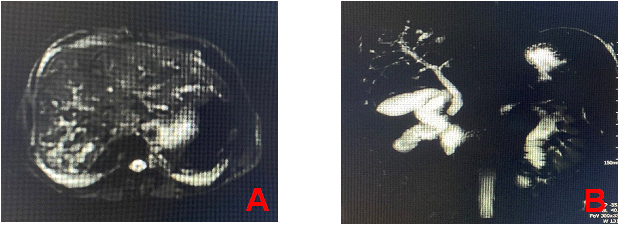
注:A. 肝内胆管走行僵硬、管壁毛躁并不均匀迂曲扩张(肝右叶胆管为著);
B. 胆囊壁增厚,胆总管下端可疑结节影呈双等信号,胆总管轻度扩张。
图1 腹部核磁共振和磁共振胰胆管造成像
肝组织活检:肝组织可见15个汇管区,肝小叶结构基本保存。汇管区显著扩大,间质大量纤维组织增生(图2A)。小胆管数目显著增多,部分胆管形态不规则及管腔轻度扩张(图2B、2C)。网状纤维染色示肝网状支架无塌陷(图2D);胶原纤维染色和天狼猩红染色显示汇管区纤维组织大量增生,局部包绕肝组织假小叶形成(图2E)。细胞角蛋白7、细胞角蛋白19、胆管上皮+,胆管数目显著增多(图2F)。 过碘酸希夫反应 +,显示肝糖原含量无增加;糖原D-PAS染色-;普鲁士蓝染色-;铜盐染色-。肝活检提示:慢性肝损伤伴胆管板畸形。轻度炎症,重度纤维化。考虑CHF。
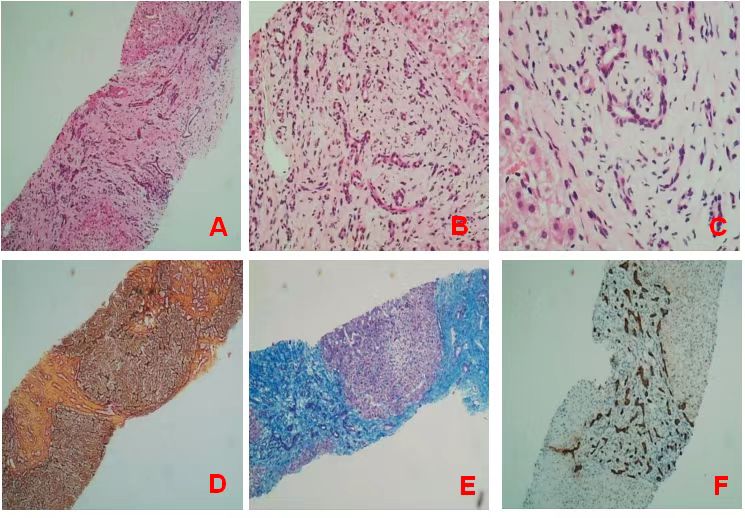
注:A. HE染色x10倍;B. HE染色x20倍;C. HE染色x40倍;
D. 网染x10倍;E. MASSON染色x10倍;F. CK7x10倍
图2 肝组织免疫组化结果
全外显子基因测序:检测到PKHD1基因变异:c.3071(exon27) T>G,p. Val1024Gly,纯合变异(图3)。ACMG评分:uncertain(PM2_Supporting+PP3)。
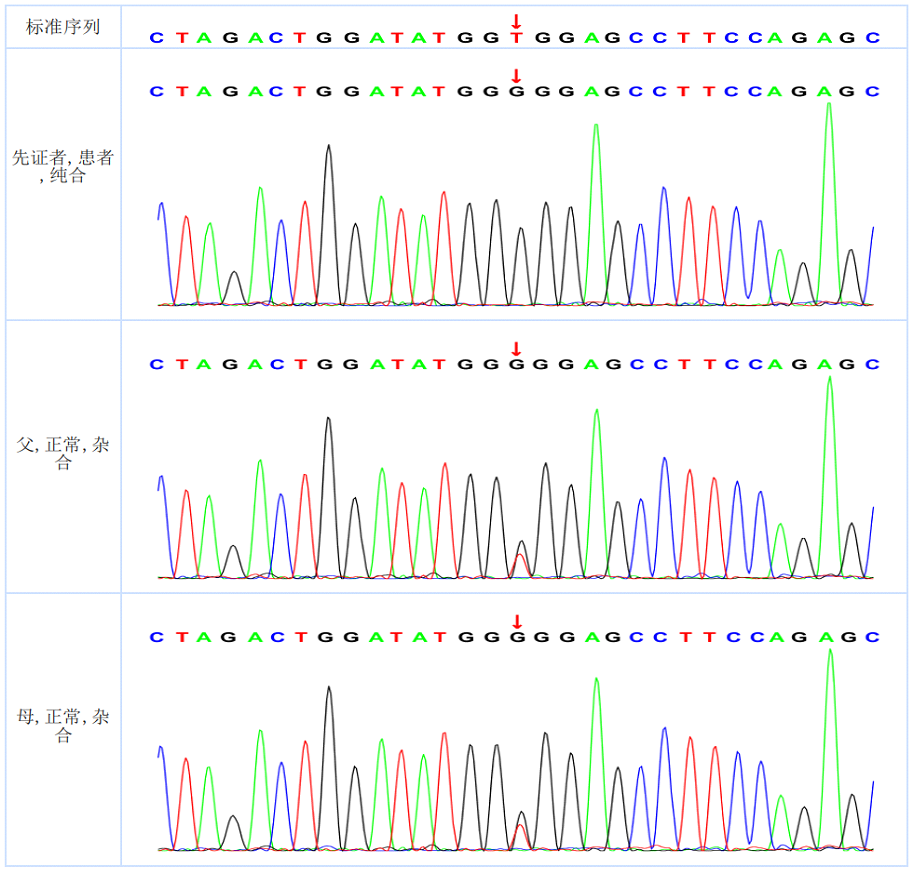
图3 基因检测结果(家系验证)
诊治经过:
入院后先予患者完善常见病因排查,后建议行肝活检。肝组织活检提示:慢性肝损伤伴胆管板畸形,轻度炎症,重度纤维化,考虑CHF。遂进一步完善基因检测,证实存在PKHD1基因纯合变异(c.3071T>G,p. Val1024Gly)。因此,该患者最终诊断为CHF(胆管炎型)。
予患者“头孢他啶”抗感染、纠正电解质紊乱、补液、舒肝利胆等对症治疗,患者体温逐渐恢复正常,复查血常规示:WBC 3.9x109/L,NE% 0.5,Hb 104g/L,PLT 377 x109/L,CRP 6.43mg/L;血沉:73mm/h;PCT:0.083ng/ml,IL-6 <1.50;生化指标以及常规止凝血指标均恢复至正常。
因基因检测结果中提示患者可能会关联多囊肾4型伴或不伴多囊肝,我们再次给予患者完善尿肾功能指标检查:N-乙酰-β-D-葡萄糖苷酶 71.6U/L(正常值:<12U/L),尿β2微球蛋白 9229ug/L(正常值:<200ug/L),尿视黄醇结合蛋白 22.2mg/L(正常值:<0.7mg/L),24h尿蛋白定量 0.61g(正常值:<0.15g),均明显高于正常值上限。虽然该患者目前未表现出肾囊肿征象,但由于存在PKHD1基因突变以及疾病进展影响,今后仍有合并肾功能损伤的风险,嘱患者定期复查检测。
3.2 供稿专家简介
张岭漪
主任医师,教授,研究生导师
兰州大学第二医院肝病科 主任
中华医学会肝病学分会、预防感染病防控分会委员
中华医学会肝病学分会肝纤维化学组委员、终末肝学组委员
中国医疗保健国际交流促进会、肝胆疾病学分会常务委员

何晶晶
兰州大学第二医院肝病科 副主任医师
中国医促会肝脏肿瘤分会青年委员
中华预防医学会感染性疾病防控分会青年委员
全国肝健康促进专家委员会委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间