
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:游绍莉
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
Shwachman-Diamond综合征(SDS)是造血功能衰竭所致不同程度外周血细胞减少的一类疾病,属于遗传性骨髓衰竭综合征(inherited bone marrow failure syndrome, IBMFS)的主要类型之一[1]。临床表现有胰腺脂肪变伴外分泌功能不全,可伴随慢性腹泻,严重时可导致营养不良;中性粒细胞减少是最常见的血液系统异常表现,部分病人会出现血小板减少;骨骼发育异常,可表现为身材矮小、骨龄落后,部分患者肋骨发育异常,此类患者往往会在儿科首诊。SDS可伴随肝脏病变,导致原因不明肝脏炎症、纤维化,因此遇到疑难肝病时应考虑该疾病。
SDS是基因突变所致的常染色体隐性遗传性疾病,90%以上是因第7号染色体的SBDS基因发生突变,目前已知SBDS基因突变有40多种。SBDS基因产物是含约250个氨基酸的多功能蛋白,主要参与细胞内核糖体的生物合成过程和有丝分裂纺锤体的稳定。SBDS基因产物广泛表达于骨髓、胰腺、肝脏和大脑等细胞代谢活跃的脏器,如出现SBDS 基因突变,将导致相关脏器功能异常。随着二代测序技术的发展普及,具有SDS样表型的其他致病基因逐渐被发现,目前已经报道包括 DnaJ热休克蛋白家族(Hsp40)成员C21(DNAJC21)、延伸因子1(EFL1)和信号识别粒子54(SRP54)。这些基因分别在核糖体亚基的组装和成熟中起作用,特别是EFL1基因突变与SDS的关系从2011年才开始逐渐被认识[2,3],目前国内尚未有相关病例报道。
SBDS通过与EFL1的相互作用,促进核糖体成熟过程中真核起始因子6(eIF6)的释放,影响80S功能性核糖体的组装(见图-1)[4,5-8],有报道EFL1 突变影响80S核糖体组装,在细胞系和动物模型中诱导SDS特征,但EFL1突变诱导SDS的精确遗传机制仍未完全阐明[5]。
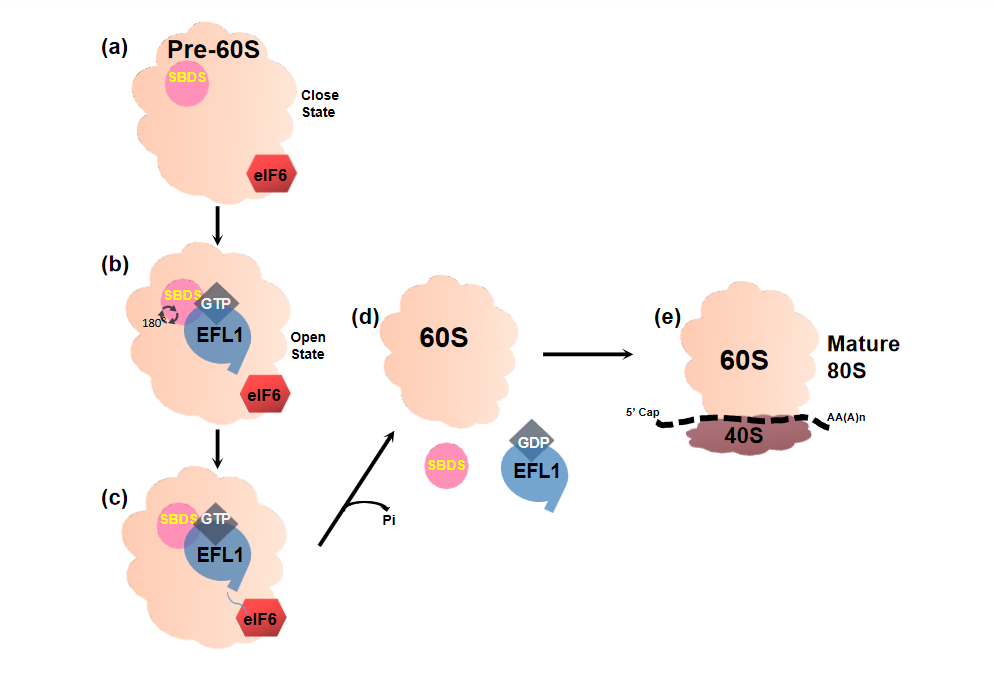
图1 SBDS和EFL1释放eIF6的机制模式图
a.前60S中SBDS处于封闭状态。b.EFL1-GTP与SBDS和eIF6结合,促进SBDS结构改变,SBDS旋转位移, SBDS转为开放状态。c.调节状态下的GTP-EFL1与eIF6竞争60S亚基上的结合位点,从而促进eIF6的位移。d.GTP水解为GDP, SBDS和EFL1-GDP与60S亚基分离。e.前60S转化为60S与40S结合,促进核糖体的成熟。
经典SDS主要临床表现包括以下几个方面[9,10]:(1)胰腺:胰腺分泌功能障碍,主要表现为慢性腹泻。本期月报中的患者(以下简称为该患者)为成年患者,表现为慢性腹泻,影像学表现为胰腺脂肪变,血清淀粉酶下降。(2)血液:主要表现为贫血、绝对中性粒细胞计数和(或)血小板减少;部分患者可发展为骨髓异常增生综合征(MDS)、急性髓细胞性白血病(AML)。该患者主要表现为明显白细胞、血小板下降。(3)骨骼及生长发育:表现为生长迟缓。该患者体瘦,伴“鸡胸”。 (4)神经系统:SDS患儿大脑双侧所有区域均出现皮质厚度增加,导致整体认知功能影响,语言及感性推理等认知水平受损等。(5)其他系统病变可能累积肝脏、心脏、胃肠道、牙齿等。研究认为约60%的SDS患者出现肝脏肿大、转氨酶升高,主要发生在婴幼儿期,随着年龄增长转氨酶趋向正常,但转氨酶正常的成人SDS患者尚可有轻度胆汁淤积表现。 目前,与EFL1基因致病性变异相关的临床表型知之甚少,有报道认为与经典SDS相比,表型可能更严重,可引起更广泛的症状[11]。
SDS的治疗主要是对症治疗。对于胰腺外分泌功能障碍,可以口服胰酶以及补充脂溶性维生素来改善胰腺功能障碍。对于造血功能异常,贫血、血小板减少者必要时可输注血红蛋白或血小板,严重的中性粒细胞减少者可使用粒细胞集落刺激因子进行治疗,而对于严重患者及骨髓异常综合征和急性髓系白血病患者,可考虑造血干细胞移植[12]。对于身材矮小,没有特效的治疗方法,同时合并生长激素缺乏的患儿,应用重组人生长因子可能有一定效果。对于肝脏受累的治疗,主要基于临床经验,转氨酶异常时可考虑保肝、降酶治疗,尽可能阻止肝纤维化、肝硬化发生。2011年国际SDS诊疗指南推荐SDS诊断标准包括临床诊断和分子生物学诊断[13]。普及开展基因检测与临床表型对比分析,根据分子机制及基因产物间的相互作用,研究基因靶向治疗成为新的方向。
本期月报报道一例因EFL1基因突变所致SDS病例,该患者因血小板较低,通过经颈静脉肝穿刺获取肝脏病理,发现病变程度相当于G1S3,纤维化明显存在,肝组织内纤维化带分隔肝实质,肝实质内炎症较轻,局部肝窦轻度扩张,肝板排列不整。这进一步加深了我们对EFL1基因突变所致该疾病肝脏病变的认识。目前该病例已经在Journal of Hepatology上发表。
参考文献:
[1] Mercuri A, Cannata E, Perbellini O,et al.Immunophenotypic analysis of hematopoiesis in patients suffering from Shwachman-Bodian-Diamond Syndrome[J].EUR J HAEMATOL, 2015, 95(4): 308-315.
[2] Wong CC, Traynor D, Basse N,et al.Defective ribosome assembly in Shwachman-Diamond syndrome[J].BLOOD, 2011, 118(16): 4305-4312.
[3] Finch AJ, Hilcenko C, Basse N,et al.Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome[J].LEUKEMIA RES, 2011, 35: S13.
[4] Bezzerri V, Cipolli M.Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives[J].MOL DIAGN THER, 2019, 23(2): 281-290
[5] Lee S, Shin CH, Lee J,et al.Somatic uniparental disomy mitigates the most damaging EFL1 allele combination in Shwachman-Diamond syndrome[J].BLOOD, 2021.
[6] Koh AL, Bonnard C, Lim JY,et al.Heterozygous missense variant in EIF6 gene: A novel form of Shwachman-Diamond syndrome?[J].AM J MED GENET A, 2020, 182(9): 2010-2020.
[7] Delre P, Alberga D, Gijsbers A,et al.Exploring the role of elongation Factor-Like 1 (EFL1) in Shwachman-Diamond syndrome through molecular dynamics[J].J BIOMOL STRUCT DYN, 2020, 38(17): 5219-5229.
[8] Tan S, Kermasson L, Hoslin A,et al.EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome[J].BLOOD, 2019, 134(3): 277-290.
[9] Valli R, Frattini A, Minelli A.Shwachman-Diamond syndrome: diagnosis, pathogenesis and prognosis[J].EXPERT OPIN ORPHAN D, 2017, 5(10): 753-767.
[10] Lawal OS, Mathur N, Eapi S,et al.Liver and Cardiac Involvement in Shwachman-Diamond Syndrome: A Literature Review[J].Cureus, 2020, 12(1): e6676.
[11] Nelson AS, Myers KC.Diagnosis, Treatment, and Molecular Pathology of Shwachman-Diamond Syndrome[J].Hematology/Oncology Clinics of North America, 2018, 32(4): 687-700.
[12] Isaev AA, Deev RV, Kuliev A,et al.First experience of hematopoietic stem cell transplantation treatment of Shwachman-Diamond syndrome using unaffected HLA-matched sibling donor produced through preimplantation HLA typing[J].Bone Marrow Transplant, 2017, 52(9): 1249-1252.
[13] Dror Y, Donadieu J, Koglmeier J,et al.Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome[J].Ann N Y Acad Sci, 2011, 1242: 40-55.

游绍莉
名师带徒学员,硕士研究生导师
解放军总医院第五医学中心肝病学部 主任医师
现任全军传染病青年委员会副主任委员
中华医学会肝病学分会重症肝病及人工肝学组副组长
中国研究型医院肝病专业委员会疑难肝病学组组长
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.综述:新兴遗传技术为Shwachman-Diamond综合征和其他遗传性骨髓衰竭疾病的个性化医疗提供信息(Blood, 2024, IF=21.0; Q1区)

2.综述:从挑战到机遇:Shwachman-Diamond综合征如何成为治疗开发的有前途的目标(Clinical pharmacology and therapeutics, 2024, IF=6.30; Q1区)
3.综述:Shwachman Diamond综合征基因型谱窄临床特征多变(Pediatric research, 2022, IF=3.10; Q1区)
4.综述:中国Shwachman Diamond综合征患者的临床及遗传学特征(Experimental biology and medicine, 2024, IF=2.80; Q2区)
5.病例报道:双等位基因 EFL1变异引起的Shwachman-Diamond综合征在婴儿早期具有复杂和致命的临床病程(British journal of haematology, 2024, IF=5.10; Q1区)
6.病例报告:Shwachman-Diamond综合征中极早期新生儿肝硬化的不寻常表现(Curēus, 2023, IF=1.0; Q3区)
7.0至18岁Shwachman-Diamond综合征患儿的生长图表(Cancers, 2024, IF=4.50; Q1区)
8.表现为儿童低血糖的代谢性肝病(Journal of clinical and experimental hepatology, 2024, IF=3.30; Q2区)
9.法国Wilson病的流行病学和经济负担:一项基于人群的全国性研究(Journal of inherited metabolic disease, 2024, IF=4.20; Q1区)
10.肝型Wilson病患者的脑形态测量(Journal of inherited metabolic disease, 2024, IF=4.20; Q1区)
三、临床资讯
3.1 病例分享:一例EFL1相关Shwachman-Diamond综合征患者
一般资料:患者女,44岁,主因血小板减少14年,腹胀、双下肢水肿1月,于2020年07月入院。
病史及体格检查:患者缘于2006年妊娠时体检发现血小板减少,具体不详,未予重视。2020年6月化验:WBC 2.6×109/L、Hb121g/L、PLT 37×109/L。AST 53U/L、ALT 62U/L、γ-GGT 24U/L,腹部CT:肝硬化、脾大、腹、盆腔积液。门诊以“肝硬化”收入我科。患者有长期腹泻病史,每日大便5-6次。流行病学史、个人史无特殊。查体:生命体征正常,身高165cm,体重45kg,BMI 16.53,营养欠佳,全身皮肤巩膜无黄染,肝掌阴性,未见蜘蛛痣。“鸡胸”表现,心肺听诊未见异常。未见腹壁静脉曲张,全腹软,无压痛、反跳痛,肝肋下未及,脾大。
入院后完善检查结果:
血常规:WBC 0.94×109/L、Hb 89g/L、PLT 29×109/L。
生化:ALT 42U/L、AST 44U/L、ALP 67U/L、Tbil 11.5μmol/L、ALB 30g/L、CHE 3424U/L、CRE 62μmol/L、淀粉酶 8U/L(正常值28-100U/L)。
其他病因学化验:PTA 48.7%,AFP 1.06ng/ml,甲功五项、肿瘤标志物均正常;血清铜、铜兰蛋白正常;自身抗体谱均阴性,IgG7.16g/L,IgM3.1g/L,ESR 7.00mm/60min;乙肝血清标志物、丙肝抗体、甲戊肝三项、丁肝三项均阴性,CMV-IgM抗体、CMVDNA阴性,血浆EBVDNA定量阴性,艾滋梅毒抗体均阴性。
影像学检查:正常范围心电图。胸部平扫未见明确异常。腹部B超:肝硬化、脾大、腹水,肝内多发稍低回声结节,脾静脉扩张。肝脏硬度值14.7Kpa。腹部血管超声:门静脉未见明显异常。腹部核磁:肝实质弥漫性损害,肝硬化,脾大,少量腹水,脾静脉曲张,脾静脉血栓形成,动脉期肝内异常信号影,考虑异常灌注;胆囊炎;胰腺显示不清,考虑脂肪变(图-2)。
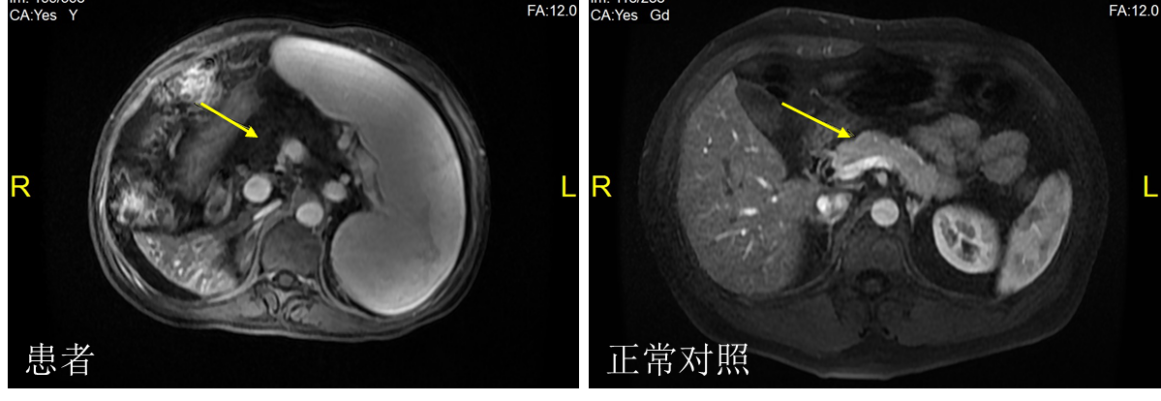
图-2 T2加权腹部核磁图(左图示患者胰腺,右图示正常人胰腺)
骨穿报告:骨髓增生活跃,粒、红两系增生,全片共见巨核细胞121个,血小板少见。
经颈静脉肝脏穿刺术:术中测肝右静脉楔压46.4cmH2O,肝静脉自由压16cmH2O,心房压12cmH2O,计算HVPG 30.4cmH2O。肝组织病理:肝组织内纤维化带分隔肝实质,肝实质内未见明显炎症坏死,局部肝窦轻度扩张,肝板排列不整。免疫组化:HBsAg(-),HBcAg(-),CK7(胆管+),CK19(胆管+),mum-1(少数+),CD34(血管+),CD10(+),CD68(散+)。特殊染色:铜染色(-),铁染色(-),D-PAS(-)。病变程度相当于G1S3,考虑非典型的肝纤维化(见图-3)。
全外显子基因测序:EFL1基因外显子区域发现两处杂合突变:①c.1581_1587delCAAATAC(母源,缺失变异),导致氨基酸改变p.K528Vfs*11(移码变异-11位后终止)。②c.74T>C(父源,胸腺嘧啶>胞嘧啶),导致氨基酸改变p.L25S(亮氨酸>丝氨酸)(见图-4)。两处位点HGMDpro数据库均未见报道。 根据ACMG指南,结合生物信息学软件预测及三维蛋白模型预测,变异位点1评级为意义未明(PM2);变异位点2评级为意义未明(PM2+PP3),且两个突变位点分别来自母源及父源,为复合杂合突变。
诊断分析:该患者临床表现为慢性腹泻、中性粒细胞及血小板低、骨骼发育异常;影像学发现胰腺脂肪变;基因检测提示EFL1基因两处突变,其中一个突变为缺失突变,另一突变位点在正常人群中携带率极低,且为复合杂合突变,符合该病遗传规律;因此考虑诊断SDS。
治疗与随访:给予患者保肝降酶、补充消化酶等对症处理后患者病情稳定出院,随访至2021年10月患者病情稳定。
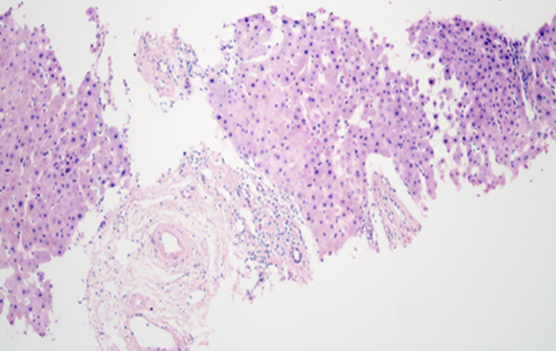
图-3 患者肝组织病理(HE染色×10)
注:汇管区扩大,纤维组织增生,纤维间隔形成,少量炎细胞浸润,未见明确界面炎。
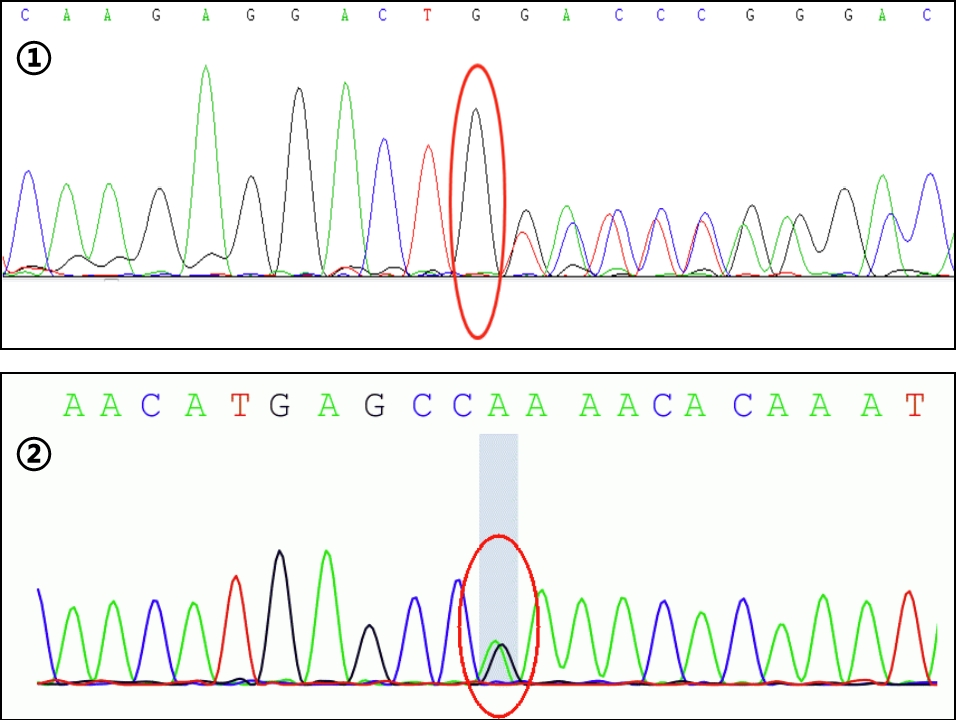
图-4 患者基因突变 Sanger 测序图
注:①chr15:82512017存在c.1581_1587delCAAATAC的杂合变异(反义链)。②chr15:82554046存在c.74T>C杂合变异。
3.2 供稿专家简介
游绍莉
名师带徒学员,硕士研究生导师
解放军总医院第五医学中心肝病学部 主任医师
现任全军传染病青年委员会副主任委员
中华医学会肝病学分会重症肝病及人工肝学组副组长
中国研究型医院肝病专业委员会疑难肝病学组组长

朱冰
副主任医师,副教授
解放军总医院第五医学中心肝病医学部肝血管疾病诊疗中心 常务副主任
北京医师协会门静脉高压症多学科诊治青年医师分会副会长
中国医药教育协会转化医学分会副主任委员
全国疑难及重症肝病攻关协作组委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间