
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:葛丽丽
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
Citrin缺陷致婴儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by citrin deficiency,NICCD)是一种因Citrin蛋白功能缺陷或不足引起的遗传性代谢性肝病,临床主要表现为胆汁淤积性黄疸、肝脏肿大、肝功能异常、生长迟缓及复杂多样的代谢紊乱,起病于新生儿和婴幼儿时期,多数预后良好,经治疗后可在1岁内缓解,少数患儿成年后可发展为成人瓜氨酸血症Ⅱ型(CTLN2),个别病例可发展为肝功能衰竭,甚至死亡[1,2]。该病分布于世界各地,以东亚地区多见,具有种族和地域差异性[2,3]。我国SLC25A13基因致病变异携带率为1/65,以长江以南地区多见[3,4]。
Citrin蛋白是一种由SLC25A13基因编码的钙结合天冬氨酸/谷氨酸载体蛋白,主要表达于肝脏线粒体内膜,参与糖代谢,尿素循环及脂肪酸的氧化分解等重要生化过程。SLC25A13基因变异可引起Citrin蛋白功能缺陷或不足,从而导致代谢紊乱、黄疸、肝功能异常、胆汁淤积等症状[5],该基因定位于人类7号染色体7q21.3,全长约200kb,由18个外显子组成,共编码675个氨基酸[6]。目前,HGMD、ClinVar公共数据库里共收录100余种致病性变异,变异类型大部分为错义、无义变异,少数为剪切和小片段缺失。该病的发病机制尚未完全明确,目前认为citrin缺陷时,一方面胞质中的瓜氨酸不能与足够的天冬氨酸反应合成琥珀酸精氨酸,导致尿素循环受阻,表现为血瓜氨酸浓度增高和高氨血症。但研究提示,瓜氨酸浓度升高不是绝对的,某些患儿该指标并未升高,可能由于其恢复较快,故早期检测瓜氨酸浓度诊断意义较大。另一方面,胞质中NADH/NAD+升高,抑制糖酵解、糖异生、尿苷二磷酸葡萄糖表异构酶,干扰蛋白质及核酸合成,促进脂肪合成,故该病患儿多伴有低血糖、半乳糖血症及脂代谢异常[7]。
NICCD患儿多于出生后数月内发病,男女比例无差异,平均出生体重低于正常[8]。患儿多因“新生儿胆汁淤积”就诊,初诊年龄为1月7 d~4月龄,平均就诊年龄为81d,实验室检查结果提示直接胆红素、总胆红素、丙氨酸转氨酶、天冬氨酸转氨酶、γ-谷氨酰胺转移酶和甲胎蛋白均明显增高[9]。NICCD临床表现和实验室检查缺乏特异性,除了胆汁淤积性黄疸、肝脏肿大、肝功能异常、代谢紊乱外,大多数患儿伴有其他合并症,如病毒性感染,发育迟滞、心脏畸形等,导致临床误诊,治疗效果不佳,病情反复而延误治疗。
有研究提出依据以下六方面进行NICCD的临床评分,并建议该评分系统可作为NICCD的疾病初筛手段,评估进一步需要做基因检测的对象。包括:(1)从未有过陶土样便;(2)门冬氨酸转氨酶/丙氨酸氨基转移酶>2;(3)直接胆红素/总胆红素<0.67;(4)碱性磷酸酶/丙氨酸氨基转移酶>10;(5)γ-谷氨酰胺转肽酶水平<300 U/L;(6)凝血酶原时间国际标准化比值>1.3。每项记为1分,NICCD患儿≥4分,其他患者≤3分,有统计学差异。但有关该病详实而明确的临床诊断标准还有待于进一步研究和证实[7,10]。目前,NICCD病主要依赖基因检测而确诊,故建议临床诊疗中,如发现新生儿期出现黄疸持续不退及肝损伤症状,需要高度怀疑该病,尽早进行基因检测,以早期识别,明确诊断。
目前,NICCD仍缺乏有效的特异性治疗方法,临床以对症治疗为主,包括饮食改善,药物治疗等。多数预后良好,少数患儿病情严重,则需要肝移植治疗,部分患儿在数年后失代偿会发展成为CTLN2,预后大多不良[10]。
综上,NICCD发病机制尚不完全明确,诊断手段仍不完善,临床诊断标准尚未建立。对于临床症状不典型的患儿,应密切关注病情变化,必要时进行基因检测协助确诊,避免误诊或漏诊。
本期月报分析了11例Citrin缺陷致婴儿肝内胆汁淤积症患儿的临床特征和基因检测结果。他们均具有典型的新生儿胆汁淤积症表现,临床初步考虑肝内胆汁淤积症可能,经过基因检查最终诊断为Citrin缺陷致婴儿肝内胆汁淤积症。通过对这些病例的复习总结,期望帮助医师尽早识别,协助临床早期精准诊断和治疗。
参考文献:
1.Zhang ZH , Lin WX , Deng M ,et al. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). PLoS One, 2014;9:e89267.
2.张婧,王晓红,叶颖子,等. 白蛋白在诊断Citrin缺陷导致的新生儿肝内胆汁淤积症的价值[J]. 中华肝脏病杂志, 2016,24(10):755-760.
3.Chen R , Wang XH , Fu HY ,et al. Different regional distribution of SLC25A13 mutations in Chinese patients with neonatal intrahepatic cholestasis[J]. World J Gastroenterol, 2013;19:4545-4551.
4.Song YZ , Zhang ZH , Lin WX ,et al. SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in East Asian patients, and the mutation distribution in a large pediatric cohort in China. PLoS One, 2013;8:e74544.
5.Balmer C , Pandey AV , Rüfenacht V ,et al. Mutations and polymorphisms in the human argininosuccinate lyase (ASL) gene[J]. Hum Mutat, 2014;35:27-35.
6.Lin WX , Zeng HS , Zhang ZH ,et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution[J]. Sci Rep, 2016;6:29732.
7.张馨怡,谭季春. Citrin缺陷致新生儿肝内胆汁淤积症的研究进展. 国际儿科学杂志,2016,43(11):884-887.
8.俞蕙,葛艳玲. Citrin缺陷导致的新生儿肝内胆汁淤积症[J]. 中华实用儿科临床杂志, 2014,29(22):1686-1689.
9.王彩红,卓志强,黄冰清,等. Citrin缺陷病6例临床特征及基因型特点[J]. 肝脏, 2020,25(11):1223-1226;1223-1226, 1240.
Chen ST , Su YN , Ni YH ,et al. Diagnosis of neonatal intrahepatic cholestasis caused by citrin deficiency using high-resolution melting analysis and a clinical scoring system[J].J Pediatr, 2012;161:626-631.e2.

葛丽丽
医学硕士
河南省儿童医院儿科医学研究所 副主任技师
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.东亚地区新生儿肝内胆汁淤积症整合单基因检测与下一代测序的诊断流程(Journal of gastroenterology and hepatology, 2024, IF=3.70; Q2区)

2.血清降钙素原可作为希特林缺陷病的诊断标志物(Clinics, 2024, IF=2.20; Q2区)
3.希特林缺陷病的发病机制和管理(Internal medicine, 2024, IF=1.00; Q3区)
4.希特林缺陷病的临床景观:多方面疾病的全球视角(Journal of inherited metabolic disease, 2024, IF=4.20; Q1区)
5.希特林缺陷病的治疗前景(Journal of inherited metabolic disease, 2024, IF=4.20; Q1区)
6.综述:希特林缺陷病(Journal of inherited metabolic disease, 2024, IF=4.20; Q1区)
7.综述:希特林缺乏症所致新生儿肝内胆汁淤积症诊断和管理的最新情况(Journal of pediatric gastroenterology and nutrition, 2024, IF=2.40; Q1区)
8.病例报道:1例希特林缺陷病患儿出现肝内多发肿块(Discover oncology, 2024, IF=2.80; Q2区)
9.病例报道:在监禁期间因饮食限制而发生的成人II型瓜氨酸血症(Internal medicine, 2024, IF=1.00; Q3区)
10.病例报道:成人II型瓜氨酸血症患者肝移植的营养支持治疗(Frontiers in nutrition, 2024, IF=4.00; Q2区)
三、临床资讯
3.1 病例分享:Citrin缺陷致婴儿肝内胆汁淤积症的临床特征和基因变异分析
研究对象:
11例NICCD患儿:男性7例,女性4例,初诊年龄在1-6月龄。
临床特征分析:
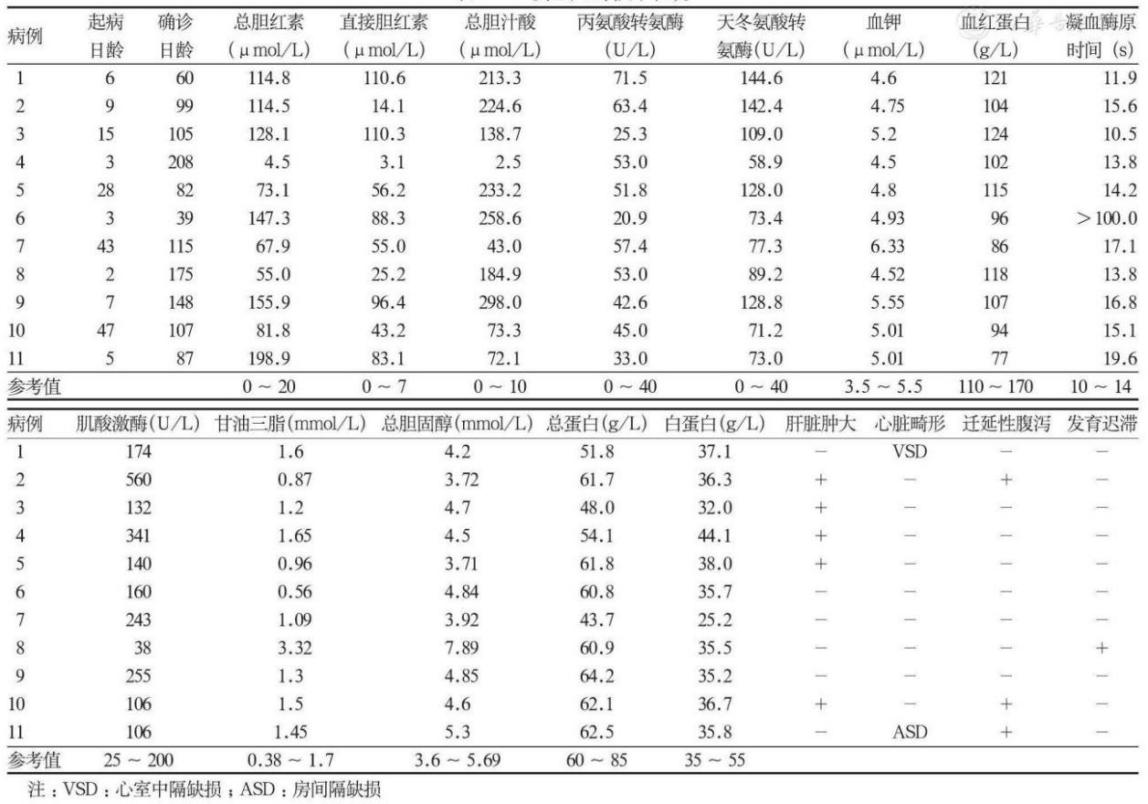
11例患儿均有不同程度的黄疸及肝损伤症状,包括肝脏肿大,肝酶异常,胆红素和胆汁酸明显升高,凝血障碍,贫血及低蛋白血症等,临床均呈现出典型的新生儿胆汁淤积症表现。11例患儿均否认家族存在遗传性疾病史,其父母们均体健,非近亲结婚。
既往研究发现大多数患儿因存在其他合并症(如贫血、病毒性感染,发育迟滞、心脏畸形等)导致临床误诊,治疗效果不佳、病情反复。在本研究中,有7例伴有凝血障碍和贫血,3例有迁延性腹泻,2例存在心脏畸形,4例心肌酶高,1例高酯血症,1例高钾血症,1例发育迟滞(表1)。
其中,病例8于出生后2天即出现皮肤巩膜黄染,合并发育迟滞、高脂血症、EB病毒感染,辗转于多家医院按“新生儿黄疸、病毒性肝炎、发育迟缓”等治疗数月,病程中先后口服“茵栀黄、葡醛内酯、熊去氧胆酸、联苯双酯、双环醇、头孢呋辛”效果差,以“发现巩膜黄染4月20天,发热1天”为代主诉入院,入院后查体:巩膜轻度黄染,腹平软,肝脾肋下未触及;血生化检查显示肝酶、总胆红素、直接胆红素、总胆汁酸升高;腹部彩超显示肝、胆囊、胆管、脾脏未见明显异常;血代谢筛查提示瓜氨酸(Cit) 、瓜氨酸Cit/丙氨酸Ala、乙酰肉碱(C2)、丙酰肉碱(C3) 偏高,疑诊为“Citrin缺陷致婴儿肝内胆汁淤积症”,遂进行基因检测,发现SLC25A13双等位基因致病性变异后确诊。
表1 11例NICCD患儿临床表现及实验室检查结果
基因变异分析:
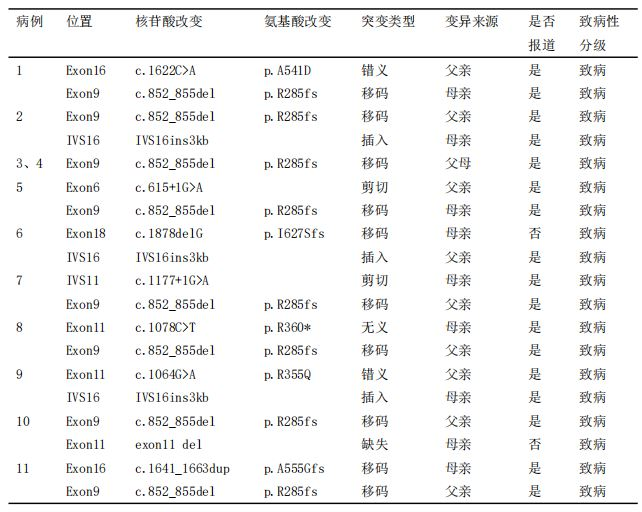
对11例患儿进行二代测序,共检测到10种不同的SLC25A13基因变异,包括3种移码变异(27%),2种剪切变异(18%),2种错义变异(18%),1种无义变异(9%),1种内含子插入(9%)。经Sanger测序和QPCR验证父母来源,所有变异均来自于父母,例3、例4为纯合变异,其余均为复合杂合变异。根据2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南对变异位点进行致病性评级,所有位点均为致病性变异(表2)。
表2 11例NICCD患儿基因变异及致病性评级
3.2 供稿专家简介
葛丽丽
医学硕士
河南省儿童医院儿科医学研究所 副主任技师
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间