
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:郑素军
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
原发性免疫缺陷病(primary immunodeficiency disease,PID)是一类由单基因变异引起免疫细胞和(或)免疫分子功能缺陷,导致机体抗感染免疫功能低下的罕见疾病[1]。PID的主要临床特点为自幼反复感染、自身炎症、自身免疫病,甚至发生恶性肿瘤。信号转导与转录激活因子1(signal transducers and activators of transcription 1, STAT1)的种系突变可导致PID,被归类为内在免疫和先天性免疫缺陷[2]。
STAT1是STAT转录因子家族的成员之一,位于人类2号染色体。STAT1基因通过选择性剪切产生两种异构体:具有转录活性的STAT1-a,编码含750个氨基酸(91kDa)的蛋白,而STAT1-b(84kDa)则因缺失部分转录激活结构域和丝氨酸727位点的磷酸化位点,发挥抑制作用。STAT1作为一种胞质蛋白,是JAK-STAT信号通路的重要组成部分,参与IFN-α/β、IFN-γ、白介素(IL)-6、IL-27、IL-10、IL-17、表皮生长因子、血管内皮生长因子、生长激素等多种细胞信号转导途径,在抗病毒和抗分枝杆菌免疫防御中至关重要[3,4]。
STAT1突变导致的PID分为常染色体显性(autosomal dominant, AD)遗传和常染色体隐性(autosomal recessive,AR)遗传。根据临床表现的不同,AD型分为STAT1功能获得型(gain of function, GOF)免疫缺陷和功能缺失型(loss of function,LOF)免疫缺陷;AR型又分为STAT1部分LOF免疫缺陷和完全LOF免疫缺陷。目前报道的STAT1突变以AD型GOF突变为主,而STAT1-LOF突变较少[5-7]。
AR遗传的STAT1完全LOF免疫缺陷,为STAT1两个等位基因均发生功能完全丧失突变,完全阻断I型(IFN-α/β)、II型(IFN-γ)和III型(IFN-λ)干扰素信号通路,临床上极为罕见[6]。已报道的突变方式包括错义突变(L600P和Q123H),小的移码缺失(c.1758_1759delAG)以及移码插入(c.1928insA)[8]。此型患者临床表现十分严重,多在婴儿期起病,常因低毒力分支杆菌或病毒(尤其是疱疹病毒如HSV、CMV或EBV)引起的侵袭性感染,往往危及生命[2]。
AR遗传的STAT1部分LOF免疫缺陷,此型患者的STAT1两个等位基因均发生功能部分丧失突变。STAT1蛋白活性降低但未完全消失。以错义突变(A46T、K201N、K211R和P696S)为主。此型罕见,严重程度较AR遗传的STAT1完全LOF免疫缺陷轻,起病年龄略晚,多为巨噬细胞内细菌感染(沙门氏菌、鸟分枝杆菌、卡介苗)和病毒感染(巨细胞病毒、水痘-带状疱疹病毒、单纯疱疹病毒1型、呼吸道合胞病毒)[9],多数可通过抗生素或抗病毒药物治愈[10]。
AD遗传的STAT1 LOF免疫缺陷:部分等位基因因磷酸化作用丧失,对IFN-γ和IFN-a的信号通路产生功能缺失效应,不能正常启动下游靶基因的转录。均为错义突变:E320Q、Q463H、K637E、M654K、K673R和Y701C。临床表现为孟德尔遗传易感分枝杆菌病(MSMD),对弱致病性分支杆菌易感,也对高毒力的结核分支杆菌易感,还会并发非伤寒沙门氏菌病感染[11]。
AD遗传的STAT1 GOF免疫缺陷:一个等位基因发生功能获得突变,使得STAT1蛋白对IFNα/β的应答能力增强。以错义突变为主,包括:R274Q、M202V、R274W、T288A、M202I、A267V。主要临床表现为慢性粘膜皮肤念珠菌病(CMC),即顽固性口腔、指甲、皮肤的念珠菌感染;合并自身免疫/自身炎症相关疾病(甲状腺功能低下、1型糖尿病、系统性红斑狼疮等),动脉瘤,肿瘤(尤其是EBV相关淋巴瘤)[10]。
STAT1免疫缺陷的治疗复杂,完全取决于突变类型(GOF或LOF)和临床表现。对于最常见的STAT1 GOF患者,核心治疗是长期抗真菌药物(如氟康唑等)预防慢性念珠菌感染[12],并联合靶向药JAK抑制剂(如芦可替尼)直接抑制过度活化的免疫通路、控制自身免疫并发症[13];当患者出现严重的自身免疫性疾病(如免疫性细胞减少症、甲状腺炎、1型糖尿病)时,可能需要使用皮质类固醇、利妥昔单抗、环孢素或其他免疫抑制剂,需警惕免疫抑制剂会增加感染风险;对于病情极度严重且药物无效的患者,可考虑高风险但可能根治的造血干细胞移植[14]。而对于较为罕见的STAT1 LOF患者,治疗核心则是预防和治疗分枝杆菌及严重感染[6]。无论何种类型,所有治疗方案都必须在专业团队的指导下进行高度个体化的制定,并辅以感染管理、免疫球蛋白替代等支持治疗。
本期月报报道一例10岁女性患儿,其自婴儿期即出现慢性、波动性肝功能异常,曾疑诊自身免疫性肝炎且对免疫抑制治疗有一定反应,同时伴有持续存在的特征性面部丘疹结节样皮疹。经基因检测发现STAT1基因致病性突变,最终诊断为原发性免疫缺陷病。慢性活动性肝炎是STAT1 突变免疫缺陷的一种较为罕见的并发症[15,16],既往也有STAT1突变可导致自身免疫肝炎的个案报道[17]。该患儿目前的治疗方案已从传统的激素和硫唑嘌呤,调整为针对该通路的新型靶向药物JAK抑制剂(芦可替尼),但近期肝功能指标仍存在波动,表明其免疫介导的肝损伤和皮肤表现尚未得到完全控制。对于此类由单基因缺陷驱动的复杂病例,其病情管理是一项长期挑战,需持续监测以评估靶向治疗疗效并防范潜在并发症。
参考文献:
1. 邢舒斌,张文静,赵晓东,等.原发性免疫缺陷病真菌易感性与机制[J]. 中华儿科杂志,2022,60(7):727-730.
2. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, Zhao X, An Y. Clinical Relevance of Gain- and Loss-of-Function Germline Mutations in STAT1: A Systematic Review. Front Immunol. 2021;12:654406. doi: 10.3389/fimmu.2021.654406. PubMed PMID: 33777053.
3. Lorenzini T, Dotta L, Giacomelli M, Vairo D, Badolato R. STAT mutations as program switchers: turning primary immunodeficiencies into autoimmune diseases. J Leukoc Biol. 2017 Jan;101(1):29-38. pii: jlb.5RI0516-237RR. doi: 10.1189/jlb.5RI0516-237RR. PubMed PMID: 27803128.
4. Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, Casanova JL. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012 Aug;24(4):364-78. pii: S0952-7915(12)00073-8. doi: 10.1016/j.coi.2012.04.011. PubMed PMID: 22651901.
5. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, Ouachée-Chardin M, Fouyssac F, Girisha KM, Etzioni A, Van Montfrans J, Camcioglu Y, Kerns LA, Belohradsky B, Blanche S, Bousfiha A, Rodriguez-Gallego C, Meyts I, Kisand K, Reichenbach J, Renner ED, Rosenzweig S, Grimbacher B, van de Veerdonk FL, Traidl-Hoffmann C, Picard C, Marodi L, Morio T, Kobayashi M, Lilic D, Milner JD, Holland S, Casanova JL, Puel A, International STAT1 Gain-of-Function Study Group. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016 Jun 23;127(25):3154-64. pii: S0006-4971(20)34434-7. doi: 10.1182/blood-2015-11-679902. PubMed PMID: 27114460.
6. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova JL. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003 Mar;33(3):388-91. pii: ng1097. doi: 10.1038/ng1097. PubMed PMID: 12590259.
7. Sampaio EP, Bax HI, Hsu AP, Kristosturyan E, Pechacek J, Chandrasekaran P, Paulson ML, Dias DL, Spalding C, Uzel G, Ding L, McFarland E, Holland SM. A novel STAT1 mutation associated with disseminated mycobacterial disease. J Clin Immunol. 2012 Aug;32(4):681-689. doi: 10.1007/s10875-012-9659-2. PubMed PMID: 22437822.
8. Le Voyer T, Sakata S, Tsumura M, Khan T, Esteve-Sole A, Al-Saud BK, Gungor HE, Taur P, Jeanne-Julien V, Christiansen M, Köhler LM, ElGhazali GE, Rosain J, Nishimura S, Sakura F, Bouaziz M, Oleaga-Quintas C, Nieto-Patlán A, Deyà-Martinez À, Altuner Torun Y, Neehus AL, Roynard M, Bozdemir SE, Al Kaabi N, Al Hassani M, Mersiyanova I, Rozenberg F, Speckmann C, Hainmann I, Hauck F, Alzahrani MH, Alhajjar SH, Al-Muhsen S, Cole T, Fuleihan R, Arkwright PD, Badolato R, Alsina L, Abel L, Desai M, Al-Mousa H, Shcherbina A, Marr N, Boisson-Dupuis S, Casanova JL, Okada S, Bustamante J. Genetic, Immunological, and Clinical Features of 32 Patients with Autosomal Recessive STAT1 Deficiency. J Immunol. 2021 Jul 1;207(1):133-152. pii: jimmunol.2001451. doi: 10.4049/jimmunol.2001451. PubMed PMID: 34183371.
9. Li C, Yin J, Li X, Yang T, Wang L, Tian Z, Zhao L, Dong H. BCG multifocal osteomyelitis. Lancet Infect Dis. 2025 Jan;25(1):e59. pii: S1473-3099(24)00584-X. doi: 10.1016/S1473-3099(24)00584-X. PubMed PMID: 39734087.
10. Martinsen KHB, Øverland T, Stray-Pedersen A, Abrahamsen TG, Fevang B, Landsverk HCE. A Norwegian cohort with STAT1-related disease - further expanding the clinical phenotype. Front Immunol. 2025;16:1620291. doi: 10.3389/fimmu.2025.1620291. PubMed PMID: 40881691.
11. Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014 Dec;26(6):454-70. pii: S1044-5323(14)00090-6. doi: 10.1016/j.smim.2014.09.008. PubMed PMID: 25453225.
12. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011 Jul 7;365(1):54-61. doi: 10.1056/NEJMoa1100102. PubMed PMID: 21714643.
13. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, Plant AS, Su HC, Allenspach EJ, Slatter M, Abinun M, Lilic D, Cunningham-Rundles C, Eckstein O, Olbrich P, Guillerman RP, Patel NC, Demirdag YY, Zerbe C, Freeman AF, Holland SM, Szabolcs P, Gennery A, Torgerson TR, Milner JD, Leiding JW. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. 2018 Nov;142(5):1665-1669. pii: S0091-6749(18)31127-8. doi: 10.1016/j.jaci.2018.07.020. PubMed PMID: 30092289.
14. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, Kim VHD, Ovadia A, Guthery SL, Pulsipher M, Lilic D, Devlin LA, Christie S, Depner M, Fuchs S, van Royen-Kerkhof A, Lindemans C, Petrovic A, Sullivan KE, Bunin N, Kilic SS, Arpaci F, Calle-Martin O, Martinez-Martinez L, Aldave JC, Kobayashi M, Ohkawa T, Imai K, Iguchi A, Roifman CM, Gennery AR, Slatter M, Ochs HD, Morio T, Torgerson TR, Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation and the Primary Immune Deficiency Treatment Consortium. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol. 2018 Feb;141(2):704-717.e5. pii: S0091-6749(17)30916-8. doi: 10.1016/j.jaci.2017.03.049. PubMed PMID: 28601685.
15. Hori T, Ohnishi H, Teramoto T, Tsubouchi K, Naiki T, Hirose Y, Ohara O, Seishima M, Kaneko H, Fukao T, Kondo N. Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism. J Clin Immunol. 2012 Dec;32(6):1213-20. doi: 10.1007/s10875-012-9744-6. PubMed PMID: 22847544.
16. Da Cunha T, Wu GY. Cytomegalovirus Hepatitis in Immunocompetent and Immunocompromised Hosts. J Clin Transl Hepatol. 2021 Feb 28;9(1):106-115. pii: JCTH.2020.00088. doi: 10.14218/JCTH.2020.00088. PubMed PMID: 33604261.
17. Hadžić N, Deheragoda M, Worth A, Bansal S, Samyn M, Kusters M. JAK Inhibition in STAT1 Gain-of-Function-Mediated Treatment-Resistant Autoimmune Hepatitis. N Engl J Med. 2024 Jan 18;390(3):284-286. doi: 10.1056/NEJMc2311867. PubMed PMID: 38231631.

郑素军
首都医科大学附属北京佑安医院,肝病中心一科主任,博士生导师
佑安肝病感染病专科医疗联盟办公室副主任
中华预防医学会感染性疾病防控分会常务委员
中华医学会肝病学分会肝炎学组 委员
中华医学会肝病学分会遗传代谢性肝病协作组 委员
中国罕见病联盟/北京罕见病诊疗与保障学会遗传性肝病分会 副主任委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.与STAT1相关疾病的挪威队列-进一步扩大了临床表型(Front Immunol. 2025,IF=5.9; Q2区)

2.具有STAT1功能获得性突变的慢性皮肤粘膜念珠菌病病例的免疫表型分析和治疗见解(J Clin Immunol. 2024,IF=5.7; Q2区)
3.具有STAT1功能获得性突变的患者显示细胞凋亡增加,而JAK抑制剂Ruxolitinib可逆转这一现象(J Clin Immunol. 2024,IF=5.7; Q2区)
4.Ruxolitinib可改善STAT1 GOF患者的免疫失调特征,但不能改善表观遗传异常(J Clin Immunol. 2024,IF=5.7; Q2区)
5.STAT-1功能获得的临床表现和治疗:来自印度的单中心经验(Pediatric allergy and immunology. 2025,IF=4.5; Q区)
6.病例报道:JAK抑制治疗STAT1功能获得介导的难治性自身免疫性肝炎(N Engl J Med, 2024, IF=78.5; Q1区)
7.病例报道:STAT1的一种新型功能丧失变体会导致孟德尔对分枝杆菌疾病的敏感性(Front Cell Infect Microbiol. 2025,IF=4.8; Q2区)
8.病例报告:两个中国兄弟姐妹的STAT1反式激活结构域出现新的新发种系功能丧失突变,其中哥哥姐姐出现多灶性卡介苗骨髓炎(Front Immunol. 2025,IF=5.9; Q2区)
9.JAG1和NOTCH2相关Alagille综合征的表型差异和疾病特异性NOTCH2变异分类指南(Liver international. 2025,IF=5.2; Q1区)
10.2012-2023年中国1431万居民罕见病谱及流行病学(Orphanet journal of rare diseases, 2025, IF=3.40; Q2区)
三、临床资讯
3.1 病例分享:原发性免疫缺陷病一例
女性患儿,10岁。因“间断肝功能异常9年余”于2025-7-15入院。
现病史:9年余前(患儿2月龄),因“百日咳、肺炎、婴儿肝炎综合征(巨细胞病毒感染)”于当地医院住院时,发现转氨酶升高,ALT/AST波动于200U/L,当地医院给予抗感染治疗后好转出院。
8年前(2岁左右)患儿体检发现转氨酶升高(最高200+U/L),当地医院查体可见颜面部红色斑疹,有融合,部分凸出皮面,不伴瘙痒,未明确诊断,予以保肝药口服。此后患儿多次因“肝功能异常、面部皮疹”就诊于当地医院,未能明确诊断。
3年余前(7岁),患儿就诊于重庆医科大学附属儿童医院,完善肝穿刺活检,肝组织病理提示:需鉴别血液回流受阻性肝病、药物/化学性肝损伤、遗传性肝病;G2S2。面部皮肤病理:轻度角化过度,角栓形成,基底层未见明显液化变性。皮损中央真皮浅中层血管及毛囊、皮脂腺周围见局灶性、小片状淋巴细胞、组织细胞浸润。PAS染色:基底膜带未见明显增厚。考虑诊断结缔组织病证据不足。基因检测提示:STAT1 c.1155_1160delGCACAC,het,likely pathogenic。诊断考虑原发性免疫缺陷病可能、自身免疫性肝炎可能,予甲泼尼龙 10mg bid、熊去氧胆酸治疗。患儿肝功能改善,面部皮疹仍未消退。此后激素逐渐减停,因肝功能波动,加用巴瑞替尼(后调整为芦可替尼)、硫唑嘌呤治疗。
近期因监测转氨酶反复,再次波动ALT 102U/L,AST 89U/L,为进一步诊治收入我科。
既往史及家族史:母亲孕期曾诊断弓形虫感染。余无特殊。
入院查体:生命体征平稳。神志清楚,面部密集粟粒至绿豆大小红色丘疹及结节,散在针尖大小白色丘疹,可见针头大小凹陷性瘢痕。全面部无破溃、渗出、鳞屑。皮肤巩膜无黄染,心肺查体未及阳性体征。腹平软,无压痛、反跳痛,Murphy征阴性,胆囊触痛阴性,移动性浊音阴性。双下肢无水肿。
入院后检查:
血常规:WBC 5.51×109/L,HGB 129 g/L,PLT 298×109/L,N% 38.8%,RETIC % 1.50%。
肝功能:ALT 31U/L,AST 30U/L, GGT 12U/L,ALP 274U/L,TBIL 19 μmol/L, DBIL 6.9μmol/L。
血常规、肾功能、凝血功能、LDH均正常。
病因学:甲肝、乙肝、丙肝、戊肝等病毒学标志物阴性;
自身免疫抗体ANA 1:100,余均阴性。特种蛋白 IgG 17.1,余阴性。
抗核抗体谱、抗线粒体M2亚型、抗中性粒细胞胞浆抗体均阴性。
肝静脉、门静脉、下腔静脉超声未见明显异常。
肝弹性 CAP 175db/m,E 3.9kPa。
腹部增强CT:獭尾肝,余腹部CT平扫及增强未见明显异常。
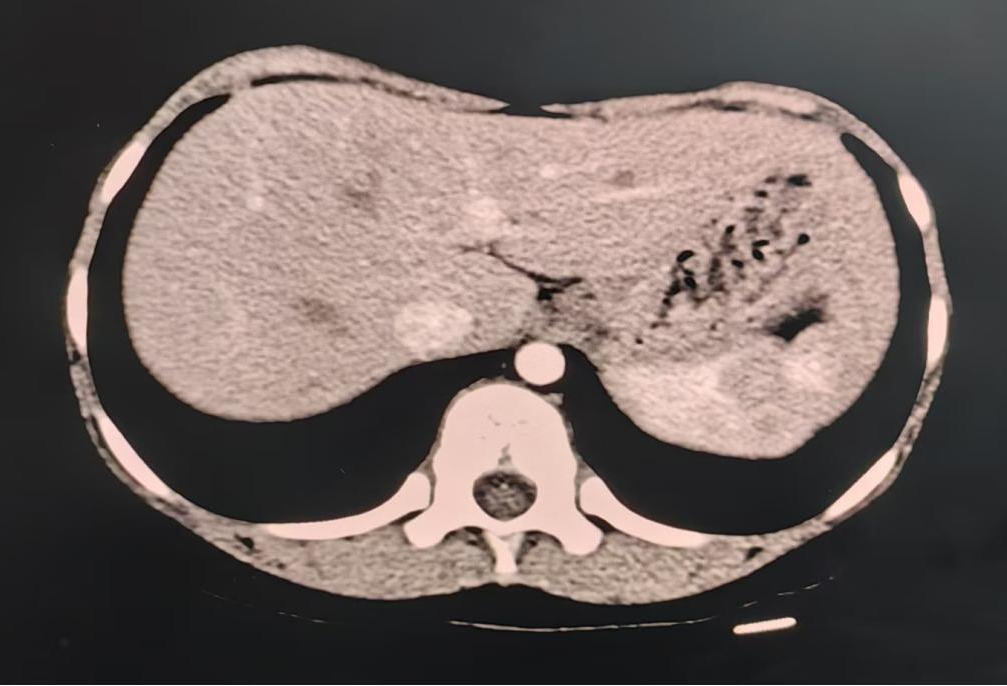
图1. 腹部增强CT,獭尾肝表现
诊疗经过:
入院后给予保肝、营养支持等治疗。请病理科会诊,肝组织病理可见汇管区纤维化扩大,淋巴细胞浸润,存在轻度界面炎;无自身免疫性肝炎病理特征。基于患儿为“STAT1功能获得性突变”,治疗核心围绕精准调控JAK-STAT通路展开:建议采用芦可替尼作为长期控制的基础治疗;同时短期联用小剂量激素快速诱导缓解。治疗的关键在于长期、动态调整药物剂量,以期在控制肝损伤及皮肤病变的同时,最大限度减少副作用。定期通过肝脏弹性检测及多学科评估监测肝纤维化进展、治疗效果及生长发育情况,实现个体化的长期管理。
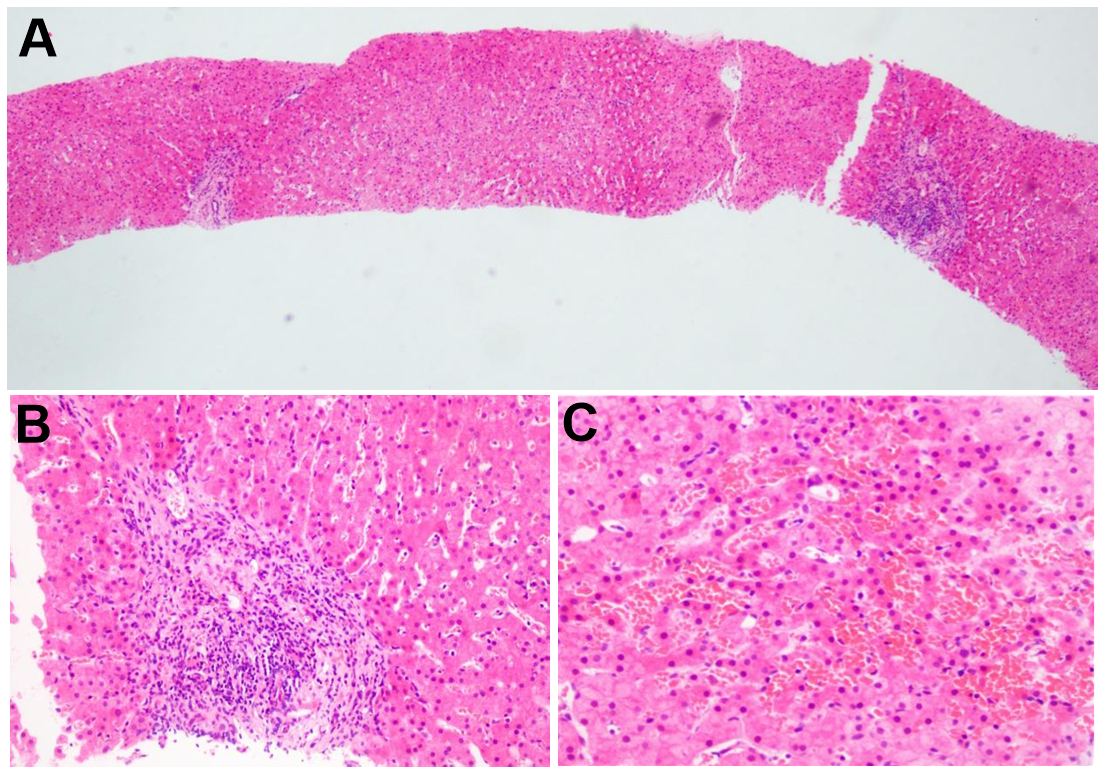
图2.肝脏穿病理。图A小叶结构存在,未见肝硬化(HEx40)。图B汇管区轻度扩大,间质轻度纤维化,间质内轻-中度单个核细胞浸润,未见明显界面炎(HEx200)。图C小叶内炎症坏死不明显,轻度肝窦扩张,局灶肝窦扩张较著,偶见巨核细胞(HEx400)
3.2 供稿专家简介
郑素军
首都医科大学附属北京佑安医院,肝病中心一科主任,博士生导师
佑安肝病感染病专科医疗联盟办公室副主任
中华预防医学会感染性疾病防控分会常务委员
中华医学会肝病学分会肝炎学组 委员
中华医学会肝病学分会遗传代谢性肝病协作组 委员
中国罕见病联盟/北京罕见病诊疗与保障学会遗传性肝病分会 副主任委员

张维
首都医科大学附属北京佑安医院,肝病中心一科,副主任医师
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-83997658
▶联系地址:北京市丰台区右安门外西头条8号佑安医院C楼7层