
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:侯维
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
遗传性球形红细胞增多症(hereditary spherocytosis,HS)是因先天性红细胞膜缺陷导致的遗传性溶血性疾病,是北欧和北美遗传性慢性溶血最常见的原因之一,发病率约1/2000[1],我国人群HS的发病率约1/10万[2]。
HS可有不同的遗传模式,以常染色体显性遗传为主,约占75%[3],15%为常染色体隐性遗传,无家族史的散发病例可能为新发生的基因突变所致[4]。目前已明确的HS致病基因包括ANK1、SLC4A1、SPTA1、SPTB和EPB42,分别编码锚蛋白、带3蛋白、α收缩蛋白、β收缩蛋白和4.2蛋白[5]。≥1个HS相关基因的突变可引起膜蛋白缺失,进而导致HS的发生[6]。研究发现,HS患者黄疸的严重程度即胆红素水平,还受参与胆红素代谢过程的关键酶尿苷二磷酸葡萄糖醛酸转移酶1家族多肽A1(uridine diphosphateglucuronosyl transferase 1 family polypeptide A1,UGT1A1)活性的影响[7]。
HS的发病机制是基因突变导致的红细胞膜蛋白缺陷,而红细胞膜蛋白是红细胞膜与细胞骨架间的垂直连接装置,对维持红细胞膜完整、维持红细胞的双面凹形状起重要作用。红细胞膜蛋白缺陷会引起红细胞膜表面积减少,使红细胞的外形由双凹圆盘状变为小球形[8],这种球形化的红细胞体积较小且僵硬,弹性降低,脆性增加,极易碎裂。因此HS患者红细胞变形能力差,不能穿越脾脏内的细小弯曲结构,无法顺畅通过脾窦,导致滞留并被脾脏破坏,红细胞寿命明显缩短,从而导致溶血性贫血、黄疸和脾大等临床表现[9]。
HS典型的临床表现为慢性溶血性贫血、黄疸和脾大。HS在任何年龄均可发病,临床症状轻重不一,可从无明显症状至危及生命的贫血、巨脾和重度黄疸。由于长期溶血,HS常并发色素性胆道结石,重者可发生胆绞痛及阻塞性黄疸。单纯HS的黄疸表现为血清总胆红素升高,以间接胆红素升高为主;而在HS合并胆管结石时,可表现为血清总胆红素升高,以直接胆红素升高为主。HS实验室检查特点为外周血球形红细胞比例增加(>10%)、红细胞脆性试验阳性[10]。诊断HS主要依据上述典型临床症状及相关实验室检查(以游离胆红素增高、网织红细胞增高,外周血涂片球形红细胞>10%,Coombs试验阴性,红细胞渗透脆性增加),如合并明确家族史,或红细胞膜缺陷分析或基因检查发现膜蛋白缺陷更有利于诊断。
全睥切除是目前HS最有效的治疗方式。脾切除虽不能改变红细胞形态,但可大幅减少红细胞破坏,使其寿命接近正常,从而改善贫血、黄疸和脾大等临床表现,是治疗HS最有效的方法[11]。HS合并有症状的胆囊结石应同时行脾切除术加胆囊切除术。
本期月报报道一例20岁遗传性球形红细胞增多症患者。患者自幼出现溶血性贫血、黄疸、脾大的表现,合并明确的家族史,存在SPTB基因突变,故明确诊断为该病。该患者反复发作胆囊炎、胆囊结石。虽然先后经过腹腔镜下胆囊切除术+胆管切开取石+胆管修补术及胆道镜下取石术,但仍反复出现胆管结石及胆道感染,且高胆红素血症无法改善。最终,该患者接受脾切除手术治疗,术后胆红素显著下降。以往有研究报道,UGT1A1酶活性降低可能导致HS患者黄疸加重,并可增加胆石症的发病率[7]。该患者合并UGT1A1基因突变,可能是加重高胆红素血症和胆石症的因素。本期月报通过分享该病例,为遗传性球形红细胞增多症患者的诊治提供了参考。
参考文献:
1.Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ; General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012 Jan;156(1):37-49. doi: 10.1111/j.1365-2141.2011.08921.x. Epub 2011 Nov 5. PMID: 22055020.
2.Wang C, Cui Y, Li Y, Liu X, Han J. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis Res. 2015 May;4(2):76-81. doi: 10.5582/irdr.2015.01002. PMID: 25984425; PMCID: PMC4428190.
3.Tole S, Dhir P, Pugi J, Drury LJ, Butchart S, Fantauzzi M, Langer JC, Baker JM, Blanchette VS, Kirby-Allen M, Carcao MD. Genotype-phenotype correlation in children with hereditary spherocytosis. Br J Haematol. 2020 Nov;191(3):486-496. doi: 10.1111/bjh.16750. Epub 2020 May 20. PMID: 32436265.
4.Manciu S, Matei E, Trandafir B. Hereditary Spherocytosis - Diagnosis, Surgical Treatment and Outcomes. A Literature Review. Chirurgia (Bucur). 2017 Mar-Apr;112(2):110-116. doi: 10.21614/chirurgia.112.2.110. PMID: 28463670.
5.Farias MG. Advances in laboratory diagnosis of hereditary spherocytosis. Clin Chem Lab Med. 2017 Jun 27;55(7):944-948. doi: 10.1515/cclm-2016-0738. PMID: 27837594.
6.He BJ, Liao L, Deng ZF, Tao YF, Xu YC, Lin FQ. Molecular Genetic Mechanisms of Hereditary Spherocytosis: Current Perspectives. Acta Haematol. 2018;139(1):60-66. doi: 10.1159/000486229. Epub 2018 Jan 22. PMID: 29402830.
7.Yi Y, Dang X, Li Y, Zhao C, Tang H, Shi X. Genetic diagnosis and pathogenic analysis of an atypical hereditary spherocytosis combined with UGT1A1 partial deficiency: A case report. Mol Med Rep. 2018 Jan;17(1):382-387. doi: 10.3892/mmr.2017.7867. Epub 2017 Oct 25. PMID: 29115431.
8.Petkova-Kirova P, Hertz L, Danielczok J, Huisjes R, Makhro A, Bogdanova A, Mañú-Pereira MDM, Vives Corrons JL, van Wijk R, Kaestner L. Red Blood Cell Membrane Conductance in Hereditary Haemolytic Anaemias. Front Physiol. 2019 Apr 16;10:386. doi: 10.3389/fphys.2019.00386. PMID: 31040790; PMCID: PMC6477063.
9.Wang C, Cui Y, Li Y, Liu X, Han J. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis Res. 2015 May;4(2):76-81. doi: 10.5582/irdr.2015.01002. PMID: 25984425; PMCID: PMC4428190.
10.周达, 陈源文, 曹海霞, 范建高. 遗传性球形红细胞增多症合并肝内胆汁淤积致严重高胆红血症1例[J]. 实用肝脏病杂志. 2015, 18(3): 310-311. DOI: 10.3969/j.issn.1672-5069.2015.02.028.
11.Vercellati C, Zaninoni A, Marcello AP, Fermo E, Fattizzo B, Giannotta JA, Bianchi P, Zanella A, Barcellini W. Changing trends of splenectomy in hereditary spherocytosis: The experience of a reference Centre in the last 40 years. Br J Haematol. 2022 Sep;198(5):912-915. doi: 10.1111/bjh.18106. Epub 2022 Mar 11. PMID: 35277856.

侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.小儿遗传性球形红细胞增多症合并胆道梗阻:16例临床分析(BMC Pediatr, 2025, IF=2.00; Q2区)

2.基于全外显子组测序技术鉴定遗传性球形红细胞增多症患者新型基因变异(Clin Chim Acta,2025,IF=2.90; Q2区)
3.遗传性球形红细胞增多症伴红细胞膜蛋白基因变异的临床特征(Front Pediatr,2025,IF=2.00; Q2区)
4.遗传性球形红细胞增多症儿童部分脾切除术后结局的基因突变相关性研究——一项多中心数据分析(J Pediatr Surg,2025,IF=2.50; Q1区)
5.先天性红细胞疾病患者患胆结石的10年风险:一项全国队列研究(American journal of hematology,2025,IF=9.90; Q1区)
6.球形红细胞增多症儿童部分脾切除术与全脾切除术相比的长期结果(J Pediatr Surg,2025,IF=2.50; Q1区)
7.综述:遗传性球形红细胞增多症诊断概述(Int J Lab Hematol. 2025, IF=2.30; Q2区)
8.综述:遗传性球形红细胞增多症概述及脾切除术治愈性效果分析(Front Physiol. 2025, IF=3.30; Q1区)
9.综述:遗传性球形红细胞增多症与胆石症的关联性研究:病理机制、诊断及临床管理方案(World J Gastrointest Surg,2025,IF=1.70; Q3区)
10.病例报道:脾动脉栓塞术作为遗传性球形红细胞增多症的主要治疗方法(Cureus,2025,IF=1.20; Q2区)
三、临床资讯
3.1 病例分享:遗传性球形红细胞增多症一例
患者女性,20岁。因“黄疸伴贫血20年,加重1月余”于2024-12-30就诊。
现病史:患者出生后出现黄疸,伴贫血、脾大,此后反复发作胆囊炎、胆囊结石。1月余前(2024-11)油腻饮食后出现腹痛,伴恶心、呕吐,就诊于北京大学人民医院急诊,完善腹部增强CT提示胆囊腔内、胆总管及左肝管内多发结石,伴肝内外胆管稍扩张,脾大,于2024-11-19行腹腔镜下胆囊切除术+胆管切开取石+胆管修补术,留置T管引流。术后出现高热,总胆红素进行性升高至890μmol/L,给予美罗培南抗感染治疗后体温好转,胆红素下降不理想,为进一步诊治收入院。
既往史及家族史:自幼诊断遗传性球形红细胞增多症;奶奶、父亲有球形红细胞增多症病史,父亲行胆囊切除术及脾切除术后好转。
入院查体:体温36.5℃,脉搏91次/分,呼吸20次/分,血压118/72mmHg。神志清,精神可,皮肤巩膜重度黄染,心肺阴性,腹软,无压痛、反跳痛、肌紧张,脾肋下10cm、剑突下3cm,移动性浊音阴性,双下肢不肿。
入院后检查:
血常规:WBC 6.04×109/L,HGB 98 g/L,PLT 122×109/L,N% 81.3%,RETIC 116.2×109/L,RETIC% 3.52%。
肝功能:ALT 108U/L,AST 143U/L, GGT 209U/L,ALP 158U/L,TBIL 829.4 μmol/L, DBIL 573.7μmol/L。
肾功能、凝血功能均正常。
病因学:甲肝、乙肝、丙肝、戊肝、细小病毒等病毒学标志物阴性,CMV DNA、EBV DNA阴性;自身免疫抗体ANA 1:100 ,余均阴性。铜蓝蛋白正常。
MRCP:胆囊切除术后,肝内外胆管轻度扩张,胆道支架术后,肝内胆管结石可能。
腹部增强CT:肝脾增大,胆囊切除术、局部引流术后改变,左肝管结石?
电子胃镜:慢性非萎缩性胃炎。
外周血全外显子基因测序:SPTB基因c.4del(p.Thr2Hisfs*44),杂合变异,被定义为疑似致病变异;UGT1A1基因c.211G>A(p.Gly71Arg),纯合变异,被定义为致病变异(图1)。

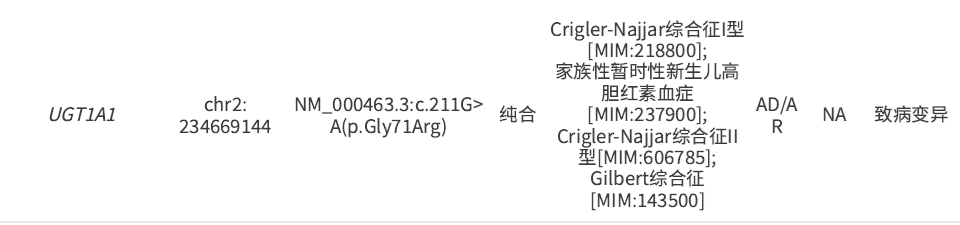
图1. 全外显子基因测序结果
诊疗经过:
入院后给予抗感染、保肝、退黄、输血、营养支持等治疗,于2025-01-04行经T管胆道造影,术中见胆管扩张,肝管内多发结石影。术后总胆红素降至371.2μmol/L(图2)。
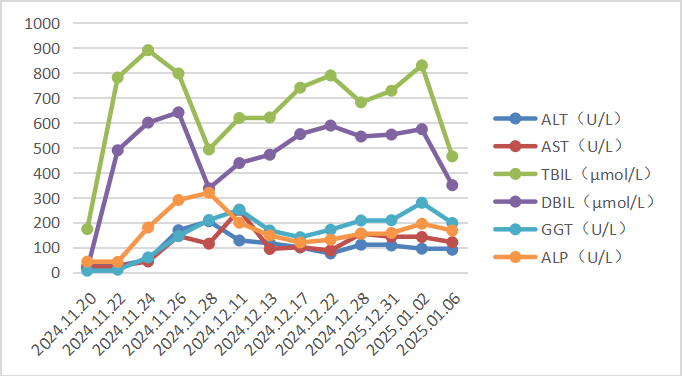
图2 胆囊切除术+胆管切开取石术后肝功能变化
此后,患者仍反复发作胆道结石及胆道感染,先后于2025-02-19及2025-03-11行胆道碎石+取石术,效果欠佳,TBIL最高升至900.3 μmol/L(图3)。
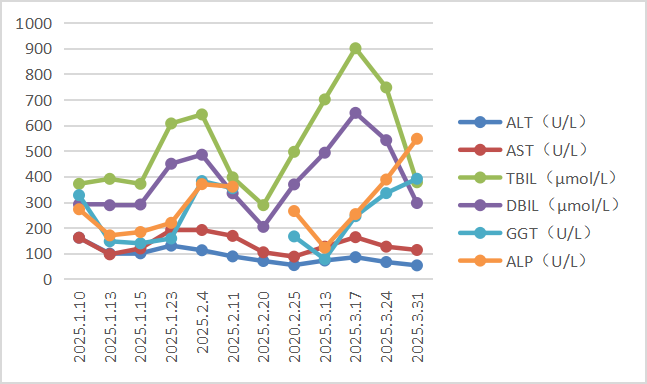
图3 胆道碎石+取石术前后肝功能变化
最终,患者于2025-04-02行脾切除术+肝活检术,术后肝功能明显改善(图4)。肝脏病理提示(图5):1.(肝组织)汇管区间质纤维化扩大,小胆管及边缘细胆管数目增多,部分管腔内见微小胆石;肝小叶结构保留,重度中心性毛细胆管淤胆,伴大量淤胆性菊形团、显著窦周纤维化及肝窦内铁沉积,并可见Kupffer细胞吞噬红细胞现象;综上,肝内重度胆汁淤积(单纯性淤胆,bland chohestasis),可见于脓毒症(sepsis)或大胆管梗阻,需结合临床鉴别。2.(脾脏)充/淤血性脾肿大,脾索增生,脾索及脾窦大量红细胞淤积,伴嗜血现象及含铁血黄素沉积,综上,结合临床符合遗传性球形红细胞增多症相关脾功能亢进。术后2月复查肝功能:ALT 53U/L,AST 45U/L, GGT 135U/L,ALP 175U/L,TBIL 47.6 μmol/L, DBIL 20.5μmol/L。
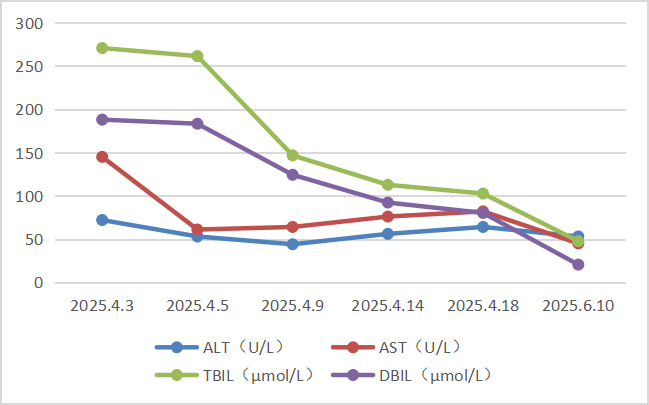
图4 胆道碎石+取石术前后肝功能变化
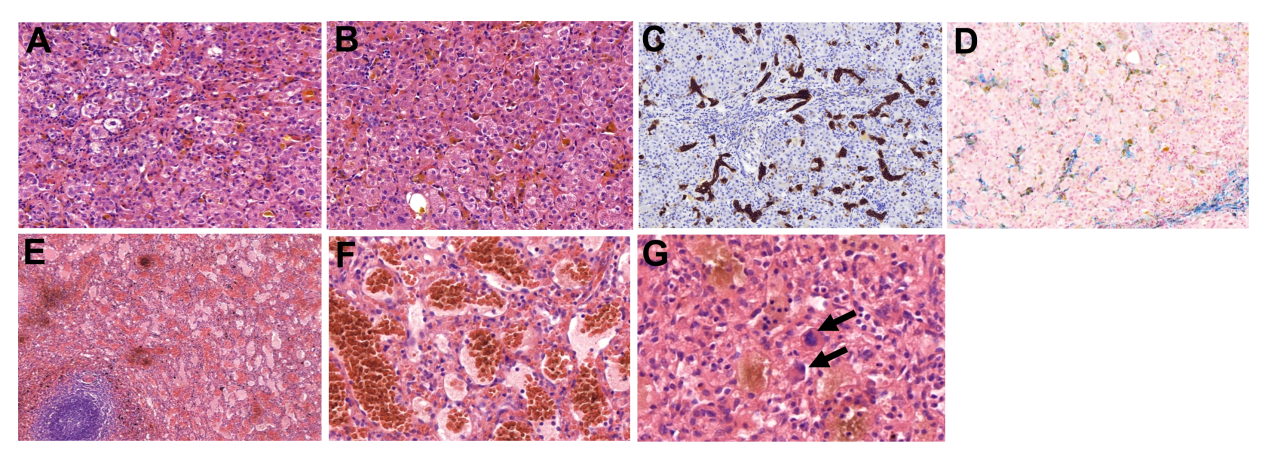
图5.肝脏病理:符合大胆管梗阻性改变;继发性铁过载。(A)小叶内广泛的毛细胆管胆栓,淤胆性菊形团形成;(B)Kuppfer细胞活化增生,吞噬胆红素;(C)汇管区扩大,轻度纤维化,边缘细胆管增生(CK7 x200);(D)Kuppfer细胞内铁沉积(Perls x200 )。脾脏病理(E、F、G)脾索、脾窦扩张充血,伴噬血现象及含铁血黄素沉积,可见髓外造血(箭头)。
3.2 供稿专家简介
侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员

王莉琳
首都医科大学附属北京佑安医院,肝病中心一科,主治医师
长期从事病毒性肝炎、肝硬化及肝癌的临床和基础研究
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-83997658
▶联系地址:北京市丰台区右安门外西头条8号佑安医院C楼7层