
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:郑素军
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
特纳综合征(Turner syndrome,TS)又称先天性卵巢发育不全综合征,属于性发育异常疾病中的性染色体异常疾病,是人类唯一能生存的染色体单体综合征[1]。发病机制是女性患者的一条X染色体完全或部分缺失,或者发生结构异常。女性中的患病率为25/100000~50/100000,占女性活产婴儿的 1/2500[2]。
Turner综合征患者在各生命周期可能发生多种并发症,并累及多个器官。临床表现多样,95-100%的Turner综合征患者身材矮小,存在卵巢早衰、子宫发育不良等原发性性腺发育不良表现,出现发育异常及先天畸形(如颈蹼,盾状胸,肘外翻,皮肤黑痣,内眦赘皮,上眼睑下垂,下颌小,第4掌骨短,指趾弯曲,股骨和胫骨外生骨疣及指骨发育不良等)。Turner综合征患者可以出现心血管畸形(主动脉缩窄,主动脉夹层,二尖瓣和主动脉瓣病变)、脊柱畸形(脊柱侧凸,脊柱后凸)、先天性泌尿系统畸形(集合系统畸形,马蹄肾,肾旋转不良)。Turner综合征患者可以合并自身免疫性疾病,其中以自身免疫性甲状腺炎最常见,此外还有糖尿病、幼年特发性关节炎、炎症性肠病等[1]。
目前Turner综合征诊断金标准是羊水细胞、外周血染色体核型分析。约半数Turner综合征患者为X单体型(45,X0),20%~30%为嵌合型(45,X0/46,XX),其余多为另一条X染色体结构异常。Turner综合征的产前诊断,常在妊娠中期超声检查发现胎儿颈部囊性淋巴瘤、全身水肿、胸膜腔积液、腹膜腔积液、颈项透明带或颈后部皮肤皱褶增厚等异常表现后,完善羊水细胞核型分析明确诊断。Turner综合征的出生后诊断,常结合生长缓慢的病史、第二性征发育不良的表型、性激素水平以及影像超声检查结果后,完善外周血染色体核型分析明确诊断。Turner综合征的治疗目的为增加患者身高,诱导性发育、维持第二性征,提高患者骨密度,同时防治各种并发症。主要包括生长激素治疗、补充性激素治疗、生育力保护治疗[1]。
约30%-50%的Turner综合征患者合并肝脏受累,并且伴随年龄增长,肝功能异常发生率增加,以GGT水平升高最常见[3]。与一般人群相比,Turner综合征患者发生肝硬化的风险增加6倍。肝脏脂肪变性、结节性再生性增生、胆道病变是Turner综合征肝脏受累的3种主要肝脏病理表现[4]。其中肝脏脂肪性变性的机制较明确,一方面由于X染色体上SHOX(矮小同源盒基因)单倍体剂量不足,导致身材矮小伴随超重,另一方面性腺功能减退及甲状腺功能减退,使Turner综合征患者易发展为血脂紊乱、胰岛素抵抗、代谢相关脂肪性肝病。Turner综合征出现肝脏结节性再生性增生和胆管损伤的机制尚不明确,可能与X染色体上存在调控血管形成基因有关,伴随X染色体缺失导致全身范围血管畸形,肝脏表现为闭塞性门静脉病、微血管异常,导致肝脏血流灌注不均匀,局部缺氧萎缩伴代偿性增生,形成结节性再生性增生等结构改变,肝内小胆管血供改变可能与胆管缺乏有关[4]。
对于肝酶升高的Turner综合征患者,不应停止雌激素替代治疗。几项研究报告了雌激素对Turner综合征患者肝功能异常的有益作用[5]。约2/3合并肝功能异常的Turner综合征患者存在胆道受累,熊去氧胆酸治疗可能对胆道病变且肝脏结构没有改变的Turner综合征患者提供有益效果[4]。
本期月报报道1例特纳综合征相关肝损伤患者,综合患者身材矮小、先天性子宫发育不良病史、全外显子测序提示一整条X染色体缺失,Turner综合征诊断明确。患者肝功能异常表现为进行性GGT升高为主,肝脏自身免疫性抗体等病因学检查均阴性,肝脏病理提示轻度脂肪肝,轻度小胆管损伤伴轻度慢性淤胆,最终考虑为Turner综合征相关肝脏受累。希望通过分享此例病例,加深临床医师对Turner综合征相关肝损伤的认识,拓展GGT升高为主肝功能异常患者的鉴别诊断思路。
参考文献:
1.特纳综合征中国专家共识(2022年版)[J]. 中国实用妇科与产科杂志,2022,38:424-433.
2.Berglund A, Stochholm K, Gravholt CH. The epidemiology of sex chromosome abnormalities. Am J Med Genet C Semin Med Genet. 2020 Jun;184(2):202-215.
3.Bourcigaux N, Dubost E, Buzzi JC, Donadille B, Corpechot C, Poujol-Robert A, Christin-Maitre S. Focus on Liver Function Abnormalities in Patients With Turner Syndrome: Risk Factors and Evaluation of Fibrosis Risk. J Clin Endocrinol Metab. 2023 Aug 18;108(9):2255-2261.
4.Roulot D. Liver involvement in Turner syndrome. Liver Int. 2013 Jan;33(1):24-30.
5..Gravholt CH, Poulsen HE, Ott P, Christiansen JS, Vilstrup H. Quantitative liver functions in Turner syndrome with and without hormone replacement therapy. Eur J Endocrinol. 2007 Jun;156(6):679-86.

郑素军
首都医科大学附属北京佑安医院,肝病中心一科主任,博士生导师
佑安肝病感染病专科医疗联盟办公室副主任
中华预防医学会感染性疾病防控分会常务委员
中华医学会肝病学分会肝炎学组 委员
中华医学会肝病学分会遗传代谢性肝病协作组 委员
中国罕见病联盟/北京罕见病诊疗与保障学会遗传性肝病分会 副主任委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.荟萃分析:肝病负担及特纳综合征相关因素(Alimentary pharmacology & therapeutics, 2025,IF=6.70; Q1区)

2.卵巢-肝脏轴:分子科学与流行病学(International journal of molecular sciences, 2025,IF=4.90; Q1区)
3.特纳综合征的代谢特征:一项真实世界队列研究(Medicina clínica, 2025,IF=2.10; Q2区)
4.门窦血管疾病:特纳综合征中未被充分认识的肝脏表现(Journal of clinical medicine, 2025,IF=2.90; Q1区)
5.特纳综合征中的肝酶升高:低度炎症和荷尔蒙失衡的作用(Journal of the Endocrine Society, 2025,IF=3.10; Q2区)
6.卵巢储备减少和早发性卵巢功能不全患者的代谢风险(Journal of clinical medicine, 2025,IF=2.90; Q1区)
7.特纳综合征患者的系统性炎症指标与肝功能障碍:一项回顾性病例对照研究(Journal of the Endocrine Society, 2024,IF=3.10; Q2区)
8.血清胆汁酸升高可预测阿拉吉尔综合征儿童肝脏预后不良:GALA 研究组结果(Liver international, 2025,IF=5.20; Q1区)
9.成人发病胆汁性肝病中全外显子测序的诊断产率(Clinical gastroenterology and hepatology, 2025,IF=12.00; Q1区)
10.脂溶刺激脂蛋白受体基因变异作为家族性肝内胆汁淤积进展性原因:病例报告(Hepatology research, 2025,IF=3.40; Q2区)
三、临床资讯
3.1 病例分享:1例特纳综合征(Turner syndrome,TS)相关肝损伤患者
患者女性,47岁,因“间断肝功能异常10年”入院。
现病史:10年前患者体检时发现肝功能异常,GGT 108U/L,转氨酶正常。平素偶有疲乏感,无厌油、纳差、皮肤瘙痒、皮肤巩膜黄染等不适症状。外院检查病毒性肝炎标志物、ANA、AMA-M2等免疫指标未见异常,B超提示轻度脂肪肝。此后患者间断复查GGT进行性升高波动于108-979U/L,ALP、ALT、AST轻度升高(ALP 105-165U/L,ALT 78-110U/L,AST 56-77U/L),伴轻度高间接胆红素血症和高脂血症。患者间断口服熊去氧胆酸治疗,效果不佳,为进一步诊治就诊于我院门诊。患者平素体健,精神、食欲、睡眠可,大小便正常,体重稳定。
既往史、个人史、家族史:甲状腺功能减退病史2年,未治疗。20余岁诊断先天性子宫发育不良、原发性闭经。否认服药史,无饮酒史。否认肝病家族史,父母及2个兄弟体健。
查体:体温36.5℃,血压120/89mmHg,身高150cm,体重60kg,BMI 26.7kg/m2,神志清楚,营养良好,皮肤巩膜无黄染,心肺查体未见异常,腹软,肝脾未触及,双下肢无水肿。
实验室检查:
肝功能+血脂(图1):
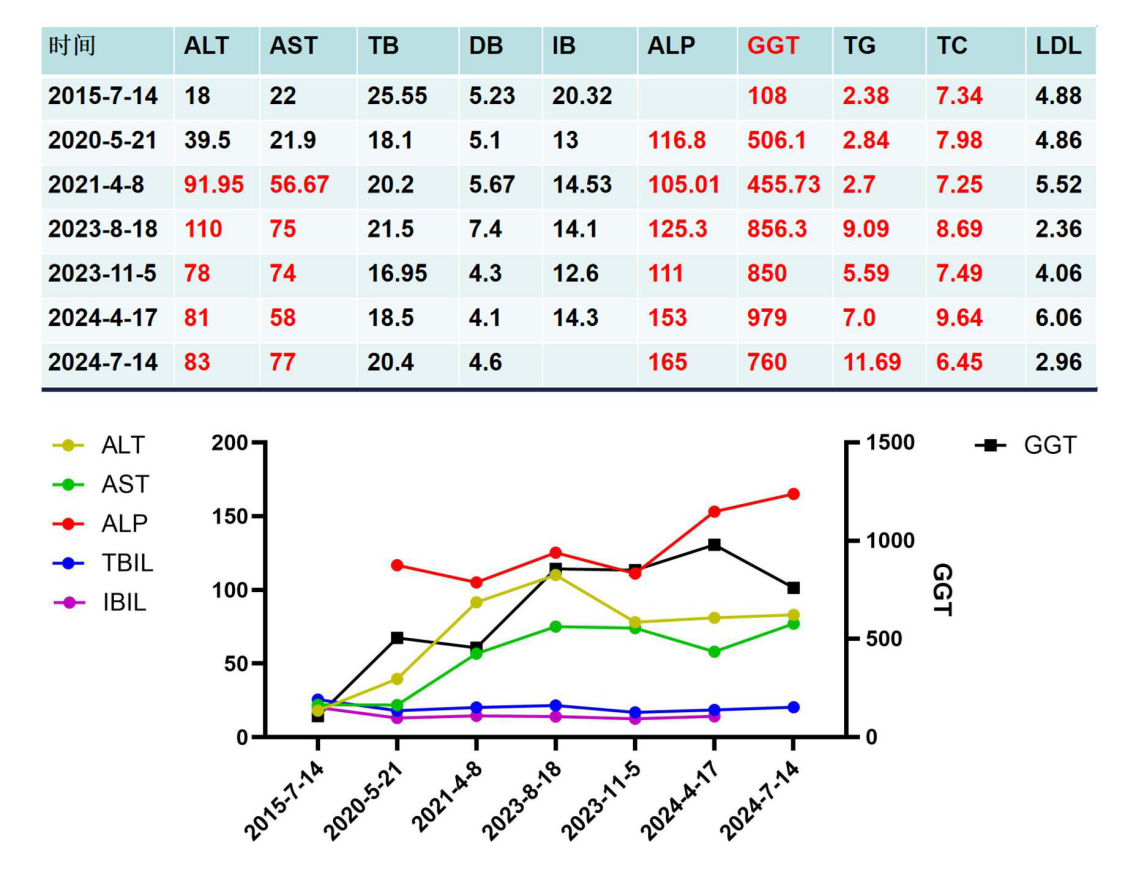
图1.患者肝功能及血脂变化
甲状腺功能及抗体:TSH 47.61uIU/mL↑,T3 1.42ng/mL,FT3 3.4pmol/L↓,T4 65.30nmolL↓,FT4 10.02pmol/L↓,甲状腺过氧化物酶抗体349↑;
一般检查:血常规、网织红细胞计数、外周血红细胞形态分析、凝血、甲胎蛋白正常;
肝脏病因检查:HBV(-),HCV(-),CMV(-),EBV(-),HIV(-),ANA(-),AMA-M2(-),gp210(-),sp100(-),ANCA(-);免疫球蛋白、铜蓝蛋白正常;
影像学检查:
腹部超声:肝右叶S6段稍高回声,肝回声增粗,轻度脂肪肝,肝内钙化灶;胆囊增大、胆囊多发结石(泥沙样)。
Fibroscan检测: CAP 302dB/m↑,E 6.3kPa;
MRCP:肝内外胆管未见明显异常;
肝穿病理: 1、小叶内未见明显炎症坏死,肝细胞空泡变性及大泡性脂变(图2.1 H&E 400x);2、汇管区轻度纤维化,间质炎症轻,多可见小叶间胆管(图2.2 H&E 400x);3、部分汇管区未见小动脉伴行小胆管(图2.3 H&E 400x);4、CK7免疫染色示汇管区未见小胆管,周围散在肝细胞呈阳性反应(图2.4 CK7 400x)。
诊断:轻度脂肪肝,轻度小胆管损伤伴轻度慢性淤胆。
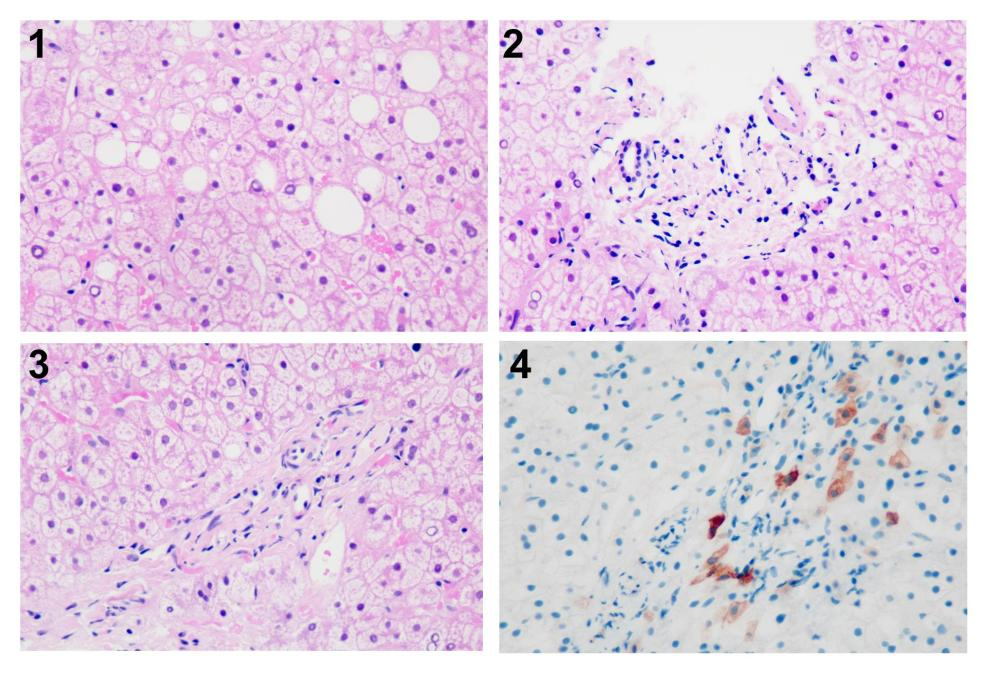
图2.肝穿病理
全外显子基因测序(图3、4):
发现与受检者临床表型相关的致病性变异:受检者在chrX:1-155270560区域检测到整条X染色体缺失,该区域在Decipher数据库中报道与Turner综合征相关(45.XO)。
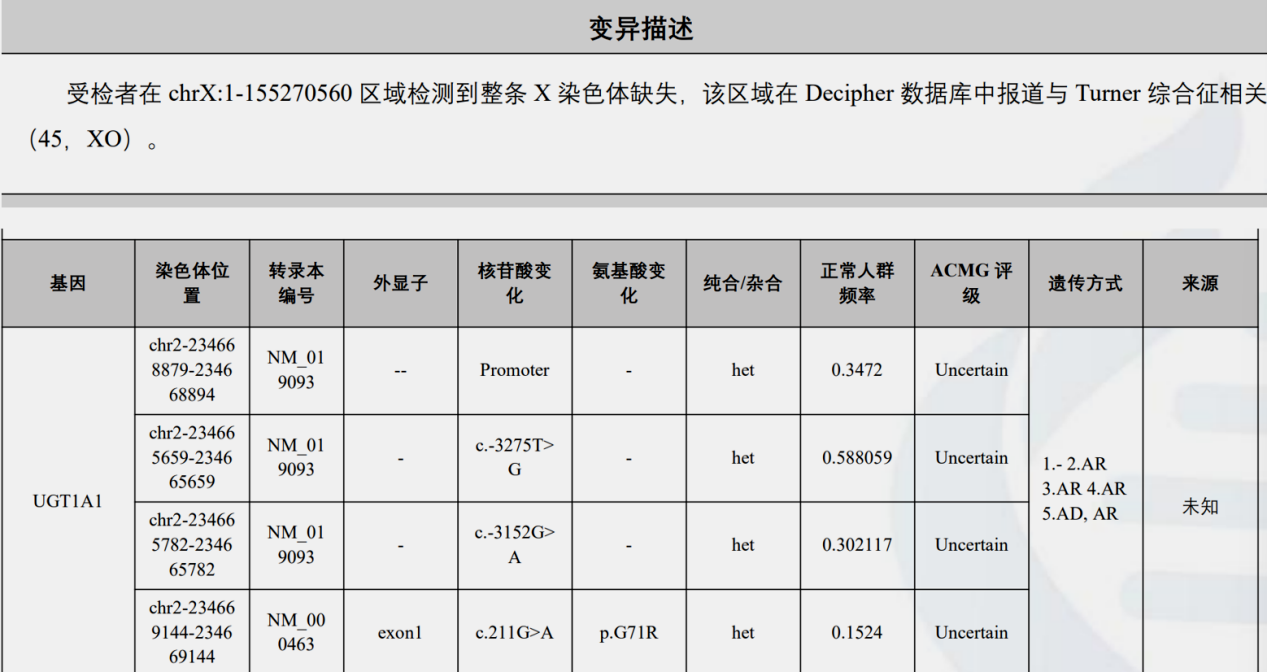
图3.全外显子测序结果
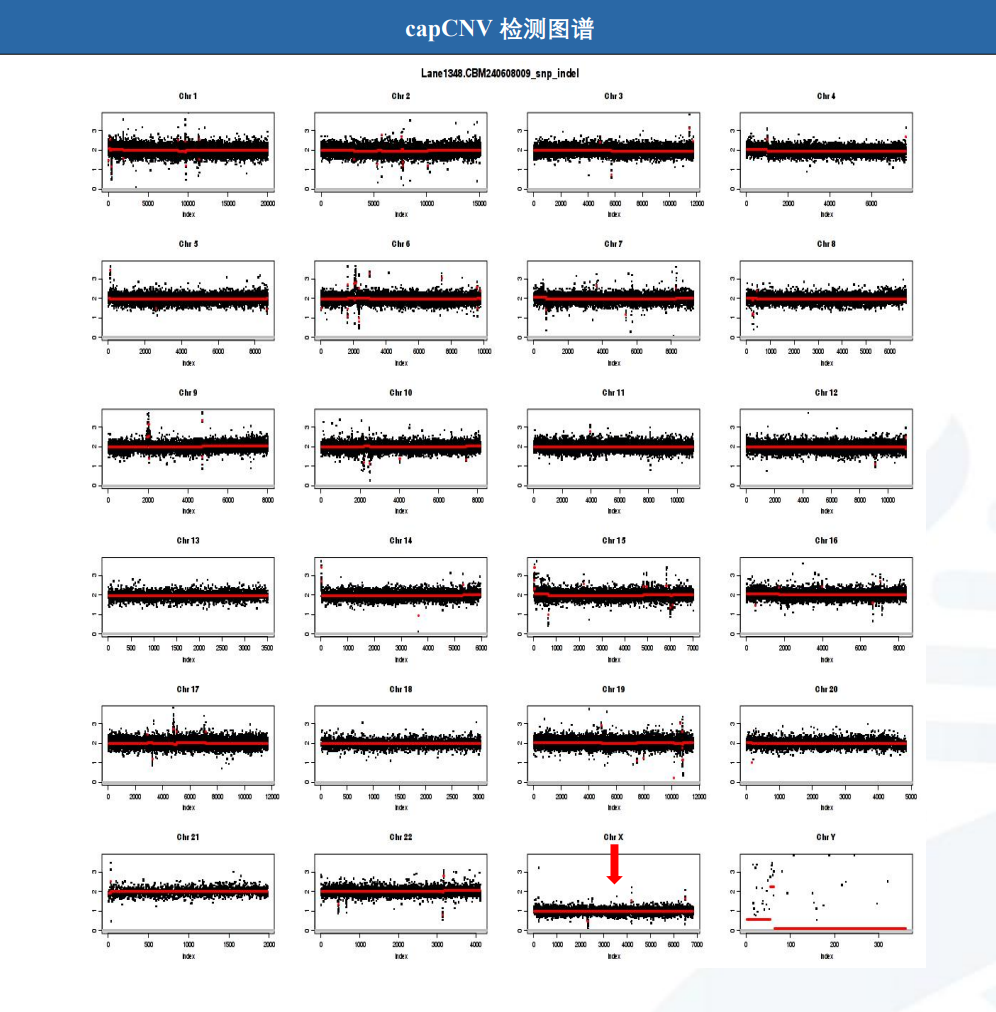
图4.capCNV检测图谱(整条X染色体拷贝数异常)
病例特点:1.中年女性,慢性病程。2.肝功能异常表现为GGT进行性升高,伴ALP、ALT、AST轻度升高,轻度间接胆红素升高,排除溶血。3.查体身材矮小、超重,有甲状腺功能减退及先天性子宫发育不全病史。4.肝脏病理提示轻度脂肪肝,轻度小胆管损伤伴轻度慢性淤胆;基因检测提示一条X染色体缺失,UGT1A1基因多位点变异。
诊断:Turner综合征相关肝损伤,桥本氏甲状腺炎,先天性子宫发育不良,Gilbert综合征,胆囊结石,高脂血症。
治疗:嘱患者低盐低脂饮食,积极运动减重。优甲乐治疗甲状腺功能减退,继续熊去氧胆酸500mg bid治疗胆汁淤积。
3.2 供稿专家简介
郑素军
首都医科大学附属北京佑安医院,肝病中心一科主任,博士生导师
佑安肝病感染病专科医疗联盟办公室副主任
中华预防医学会感染性疾病防控分会常务委员
中华医学会肝病学分会肝炎学组 委员
中华医学会肝病学分会遗传代谢性肝病协作组 委员
中国罕见病联盟/北京罕见病诊疗与保障学会遗传性肝病分会 副主任委员

於海天
首都医科大学附属北京佑安医院内科学博士
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-83997658
▶联系地址:北京市丰台区右安门外西头条8号佑安医院C楼7层