
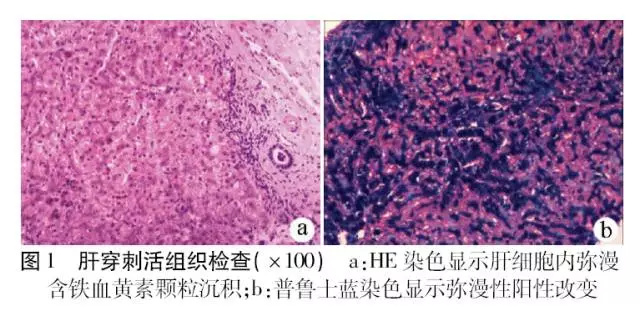
患者男性,32岁,因“反复转氨酶升高5年余”于2016年5月入院。查体:面色青黑,其他皮肤褶皱处色素沉着,脾大,肋下3 cm可触及,心肺听诊未见明显异常。实验室检查:ALT 122 U/L,AST 49 U/L,Alb 43.3 g/L,空腹血糖5.2 mmol/L,血清铁蛋白(SF) 7614 ng/ml,血清铁(SI) 201 μmol/L,总铁结合力(,TIBC) 279 μmol/L,转铁蛋白饱和度(TS)72%;嗜肝病毒和非嗜肝病毒、甲状腺功能、血常规、尿常规检测均阴性。彩超:肝实质回声致密增强,脾脏增大。患者无肝活组织检查禁忌,与患者沟通后签署知情同意书,行肝穿刺活组织检查:肝细胞内弥漫性含铁血黄素颗粒沉积,肝小叶灶状坏死,伴巨噬细胞聚集并吞噬含铁血黄素颗粒,部分小胆管上皮细胞内可见含铁血黄素颗粒,普鲁士蓝染色肝细胞内弥漫性阳性,部分胆管上皮细胞及吞噬细胞阳性,符合HH表现(图1)
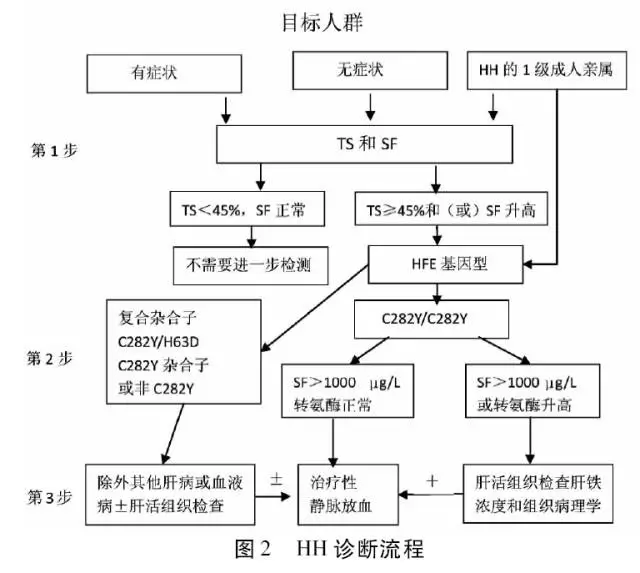
诊断标准参考2011年版《美国血色病诊疗指南》(图2),结合患者临床表现、实验室检测和病理结果明确诊断为HH。为进一步明确其遗传背景,在告知并签署知情同意书后,采集患者及其他5位家属外周静脉血,离心后分离血清、血浆,并将样本置于-80 ℃冰箱保存,用于基因检测。
先证者及家系成员一般资料及铁代谢相关检测结果见表2(略)。
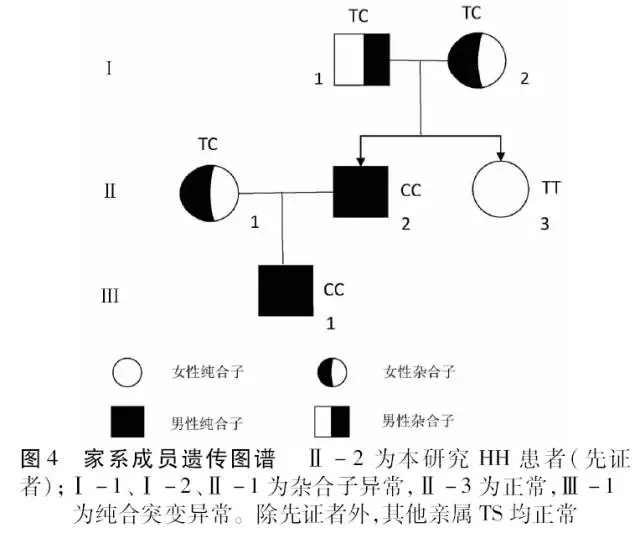
先证者血色病相关HFE、HJV、HAMP、 TfR2、SLC40A1基因检测中,仅在HFE基因外显子EXON2 相应区间序列2号内含子第4个碱基出现T→C纯合突变(IVs 2+4T→C, C/C纯合,splicing,异常)。患者儿子与先证者同为HFE IVs 2+4T→C纯合突变,患者父母和妻子均为IVs 2+4T→C, C/T杂合突变,患者妹妹为正常TT纯合表型(图3)。
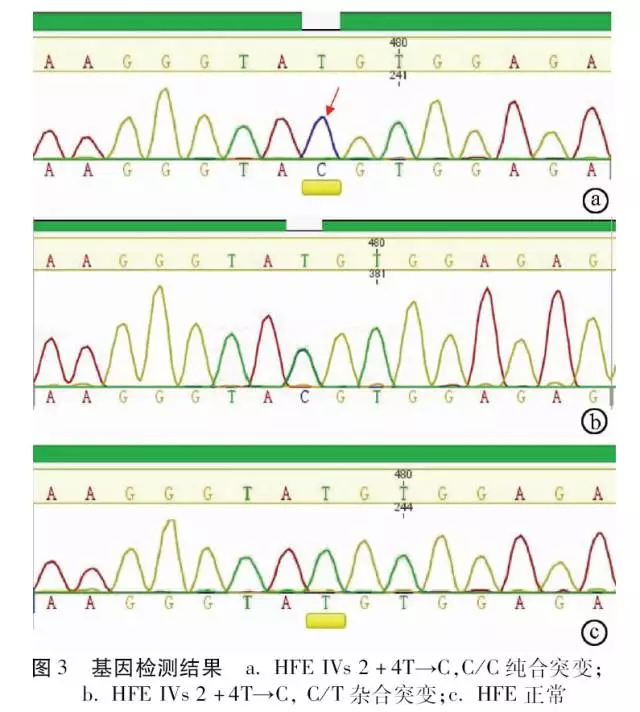
根据测序结果, 明确每位家系成员的HFE基因型, 以IVs 2+4T→C等位基因为基础,绘制家系图谱(图4)。
随着基因检测技术的不断发展,血色病的诊治有了飞速的进展。目前认为HH的发病机制可能与HFE、HJV、HAMP、TfR2和 SLC40A1等基因突变有关,导致转铁蛋白-转铁蛋白受体机制紊乱。在国外,HFE基因异常尤其是C282Y位点的基因突变与80%表1HFE /HJV/HAMP/TfR2/SLC40A1基因引物序列的HH患者有明确的相关性,被认为是诊断HH的有力依据。在我国HH基因突变主要为个案报道,仅发现HJV C321X、HFE H63D和HFE IVS 3+5T>C等少数非典型HFE突变,然而其遗传机制尚不明确。HFE 是第一个被确定的血色病相关基因,其中C282Y(酪氨酸取代282位半胱氨酸)和H63D(天冬氨酸取代63位组氨酸)是最主要的2个致病性突变位点。在欧美国家,尤其是在高加索人群中,约85%~90%的HH患者带有C282Y纯合突变或C282Y/H63D复合突变,然而HFE C282Y基因突变在亚洲人群中较低。这也提示亚洲人群中HH的遗传基因或以非HFE 相关性血色病为主。因此应更关注非HFE的突变类型,以期发现中国HH的基因遗传背景。
在线人类孟德尔遗传数据库根据不同基因突变情况将 HH 分为 4个类型:1 型为 HFE 突变所致,故又称为 HFE 相关血色病;2 型为 HJV 基因(2A)和 HAMP 基因(2B)突变所致;3 型为 TfR2 基因突变所致;4型为 FPN 基因突变所致。HJV mRNA 主要在肝脏、心脏和骨骼肌中表达,其中肝实质细胞内 HJV的表达对铁调素的分泌具有重要的调节功能,进而维持机体内铁代谢的平衡,其异常突变导致铁调素表达降低而致病,其中p.C321X突变类型在我国多次报道,此外p.I281T 纯合突变、p.R326X突变也被证实可引起 HJV 蛋白缺失尾部的糖基磷脂肌醇锚定结构域,进而形成可溶性HJV,最终引发铁代谢异常,被认为是我国最常见的基因突变类型。HAMP 编码的是肝脏分泌的小分子肽类激素,是负性铁调节激素。HAMP 基因本身缺失的表型与HJV功能缺失导致的表型一致,其铁蓄积严重且速度更快。TfR2 基因突变是在2000年发现的常染色体隐性遗传形式。它位于染色体7q22,由18个外显子组成,编码TfR2蛋白,参与肝细胞摄取转铁蛋白结合铁,并与 HFE 结合后,调节铁调素的表达从而致病[12-13]。FPN基因突变引起的铁超负荷与前几种基因不同。FPN 基因包括2种突变类型:一种是突变导致其编码的铁泵蛋白不能定位在细胞表面的“功能缺失性突变”,铁离子主要沉积在网状内皮系统 (肝和脾),此种情况被称为膜转铁蛋白疾病;另一种是表达于细胞表面的FPN产生铁调素抵抗“功能获得性突破”,与HFE 相关血色病类似。
本研究的HH家系中,先证者及其儿子属于HFE基因突变,其表型为2号内含子第4个碱基出现T→C纯合突变(IVs 2+4T→C, C/C 纯合突变),患者妻子和父母表现为该表型的T/C杂合突变,其妹妹为正常纯合表型,该遗传形式符合遗传定律,属于常染色体的隐性遗传。该家系先证者发病,SF、TS、SI、TIBC均明显升高,肝活组织检查可见明显大量铁沉积,诊断明确。此外,指南中提出HH按病程可分为3期: 1期为患者具有“遗传易感性”, 但尚未发生铁过度沉积;2期为患者具有铁过度沉积的显型证据, 尚无组织或器官损伤; 3期为患者铁过度沉积, 导致组织和器官损伤。患者儿子存在同样基因突变但尚未发病,处于疾病的1期阶段,支持除遗传因素外,多种因素所致患者体内铁过载进而引起相应器官损伤而致病。本研究发现的基因突变在我国尚未见报道,这或许是我国HH患者发病相关的基因表型,但其影响基因突变相关氨基酸或蛋白质功能的变化及具体发病机制仍需要大量研究进一步证实和探讨。鼓励HH相关基因突变的新类型个例及家系的报道,积极开展有关血色病的大规模人群流行病学调查,确立与中国人群相关的 HH 基因突变类型库,通过快速、有效的临床基因诊断方法对疾病做出早期诊断及防治是HH研究的重点,这也是本病未来诊治的基础。
节选自
宁会彬, 何佳, 李俊利, 等. 1例携带HFE基因剪切突变的遗传性血色病患者家系调查[J]. 临床肝胆病杂志, 2017, 33(1): 155-159.
朋友会在“发现-看一看”看到你“在看”的内容
确定
已发送
朋友将在看一看看到
确定发布到看一看
确定发送中
微信扫一扫
关注该公众号
使用小程序