
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:许红梅
执行编辑:郑素军,汤珊,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
糖原累积病 (glycogen storage disease, GSD) 是一组糖原分解或合成代谢功能障碍引起的疾病,主要是相关酶缺乏引起。Ⅱ型糖原累积病 (glycogen storage disease, typeⅡ, GSDⅡ) 是一种由于酸性α-葡萄糖苷酶 (acidα-glucosidase, GAA) 基因突变导致的常染色体隐性遗传性代谢性疾病, 也称为Pompe病(庞贝病)。1932年由荷兰病理学家提出。GSDⅡ发病率低, 该病白种人发病率为1/40 000,汉族人发病率1/18000。其发病主要是因为编码该酶的GAA基因突变所致, GAA基因突变造成酶活性下降或缺陷, 导致进入溶酶体的糖原无法被分解而持续堆积, 溶酶体增生或破坏, 导致细胞结构及功能损害, 从而导致心肌、骨骼肌、肝脏、脾脏、血液系统、神经系统及血管等病变。
该病有一系列表型,每种表型都包括肌病,但发病年龄、器官受累和临床严重程度有所不同。临床上根据发病年龄主要分为3型, 婴儿型 (<1岁) 、青少年型 (1~19岁) 和成人型 (≥20岁) 。婴儿型又称早发型,后两者为晚发型。
婴儿型为婴儿在出生后几周到几个月内出现肌张力减退、全身性肌无力并出现“婴儿软弱无力”症状、神经病变性延髓无力、喂养困难、巨舌症、肝肿大和肥厚性心肌病,通常在1岁内死于心脏呼吸衰竭或呼吸系统感染。青少年型以四肢无力为主要临床表现,逐渐进展出现呼吸困难、发绀、心脏扩大、心力衰竭及腓肠肌肥大,病情进展较慢,常因肺部感染致呼吸衰竭而死亡。但部分患者可生存20年以上。成人型为30~40岁发病,缓慢进展性的四肢肌肉萎缩、无力,近端较远端重;以躯干肌、骨盆带肌明显,半数以上患者影响呼吸肌,预后相对较好。
GSDⅡ临床症状轻重主要与患者体内的GAA残余酶活性有关。在婴儿型患者,GAA活性通常完全缺失或小于正常值的1%;在晚发型患者体内残余的GAA活性稍高, 但常小于正常值的30% (2%~30%) ;对于酶活性大于正常值40%的患者,临床上多无相关临床表现。
本期月报报道1例3岁儿童糖原累积病Ⅱ型的临床病例,该病例最初行走乏力、步态摇摆、上楼梯困难,肝功能检查异常。经基因检测后最终诊断明确。通过对该病例的分析,以期临床医生提高对GSD II的认识,对于原因不明的肝功能损害,需要拓宽思路,积极进行相关基因检测,警惕遗传代谢疾病等罕见病存在。

许红梅
主任医师,博士生导师
重庆医科大学附属儿童医院感染学科主任
中华医学会儿科学分会感染专业学组副组长
中华医学会感染性疾病分会儿童感染及肝病学组副组长
中华医学会儿科分会儿童医院感染控制委员会副主任委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
■综述:威尔逊病铜转运蛋白ATP7B的结构(Sci Adv,2022, IF=14.136; Q2区)

■威尔逊病的脑磁共振成像和神经系统疾病的严重程度相关(Neurol Sci,2022, IF=3.307; Q3区)
■血清铜蓝蛋白对儿童威尔逊病的诊断价值评估(BMC Gastroenterol,2022, IF=3.061; Q3区)
■中国威尔逊病患者的临床和遗传特征(Transl Neurodegener,2022, IF=8.014; Q3区)
■血清神经丝轻链可作为威尔逊病脑损伤的生物标志物(Mov Disord,2022, IF=10.338; Q2区)
■生物阻抗相位角可作为迟发性庞贝病的预后工具:一项 15 年随访的单中心前瞻性研究(Front Cell Dev Biol,2022, IF=6.684; Q3区)
■综述:庞贝病基因治疗进展(Biomedicines,2022, IF=6.081; Q3区)
■晚发型庞贝病的早期临床表型(Mol Genet Metab,2022, IF=4.797; Q3区)
■综述:通过调节线粒体-溶酶体轴治疗溶酶体贮积病(Cells,2022, IF=6.6; Q3区)
■血清代谢组学分析显示吉尔伯特综合征的脂质分解代谢增加(Metabolism,2021, IF=8.694; Q3区)
三、临床资讯
1.病例分享
现病史:患儿男, 3岁, 因“发现肝功能异常4个月”于2016.4.3入院, 入院前4个月患儿家属发现患儿行走乏力、步态摇摆、上楼梯困难于当地医院住院,常规检查肝功能“轻微异常”, 诊断病因不清,给与口服谷胱甘肽。半月前复查肝功能仍异常,故转我院进一步诊治。
生长发育史: 追问病史,患儿1岁3月开始扶走, 逐渐可独走,但行走后乏力,上楼梯较困难,即患儿大运动发育落后。
孕产史:患儿系G2P2, 孕40周, 出生体质量3.4 kg。母亲否认流产史,家族中无早夭儿童、无类似疾病患者等病史。G1P1, 女, 10岁, 体健。
入院后查体:神智清晰,反应好,无上睑下垂,舌大不明显。双肺呼吸音清晰,呼吸平稳,无发绀,无三凹征,心率116次/分,心律齐,心音有力,肝肋下3 cm, 剑突下3 cm, 质中, 边缘锐, 表面光滑, 腓肠肌质地偏硬但无明显肿大, 四肢肌力、肌张力未见异常。
辅助检查:2016年2月4日肝功能(当地医院):ALT96 U/L, AST 103 U/L, ALP 268 U/L。2016年3月21于本院门诊肝功能:ALT 84.4U/L, AST 83.7 U/L, LDH 346.4 U/L。2016年4月3日住院后肝功能:ALT 82.2 U/L, AST 81.1 U/L, LDH 479.0 U/L。心肌酶谱:肌红蛋白117.5μg/L↑, 肌酸激酶497 U/L↑,肌酸激酶MB同工酶 10.2μg/L↑,α-羟丁酸脱氢酶420 U/L↑,肌钙蛋白正常。腹部彩超示肝脏稍肿大, 其内结构未见明显异常, 脾脏未见明显异常。粗大运动评价:实际运动年龄相当于21月龄, 精细运动评价相当于30月龄小儿水平。肌电图正常。因肌活检为侵袭性,家长拒绝。
基因检测:本例患儿检测到2个基因突变位点:①c.2238G>C(p.W746C),为错义突变,来源于其父亲,突变位点位于16外显子;② c.2608C>T(p.R870X),为无义突变,突变来源于患儿母亲,突变位点位于18外显子。测序图如下:
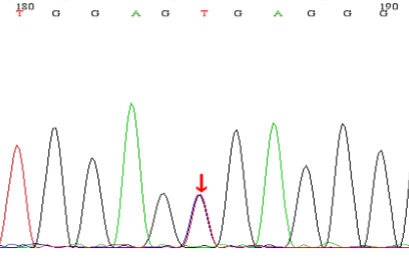
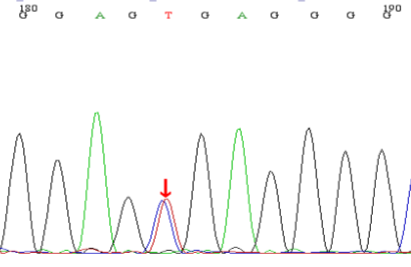
图1:患儿及其母亲基因测序结果:
A为患儿c.2608C>T杂合突变,
B为患儿母亲c.2608C>T突变携带者
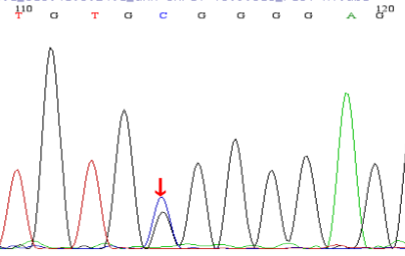
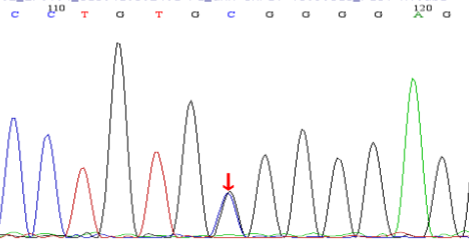
图2:患儿及其父亲基因测序结果:
A为患儿c.2238G>C突变;
B为患儿父亲为c.2238G>C突变携带者。
综上所述,本例患儿以运动发育迟缓、行走乏力为表现,肝大,肝酶异常、心肌酶异常,通过基因组测序发现患儿有来源于其父亲及母亲GAA基因突变,进而确诊为GSD II。该患儿起病隐匿,病程迁延,临床表现不十分典型,临床容易出现误诊或延误诊治等情况,对于早期行全基因检测可能对疾病早期诊断及治疗具有一定帮助。
治疗:GSD II治疗主要包括低糖高蛋白或支链氨基酸饮食、心肺功能支持、康复训练等。阿葡糖苷酶α对于GSDII为特异性治疗,可延缓甚至阻止病情进展,改善运动、呼吸功能等,改善患者生活质量,该药已经上市。本病例患儿基因确诊后因当时酶替代药物不可及未行特异性治疗。
1.2供稿专家简介
许红梅
主任医师,博士生导师
重庆医科大学附属儿童医院感染学科主任
中华医学会儿科学分会感染专业学组副组长
中华医学会感染性疾病分会儿童感染及肝病学组副组长
中华医学会儿科分会儿童医院感染控制委员会副主任委员
四、联系方式
投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
联系电话:010-63291007
联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间