
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:李新华
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
糖原贮积症(Glycogen storage disease,GSD)是一组罕见的遗传性酶缺陷导致的糖原代谢紊乱性疾病,存在糖生成、糖原分解、糖酵解或葡萄糖转运等代谢障碍,其中多数是糖原分解酶缺乏导致糖原在组织内过度沉积。根据所缺乏酶的种类不同和糖原在体内沉积的组织不同,糖原贮积症有多种不同类型,OMIM数据库记录的临床类型至少在16种以上,除了IXa/d型为X连锁隐性遗传外,其余各型均为常染色体隐性遗传。
GSD多发于婴幼儿和青少年,临床表现复杂多样,可累及机体各个组织器官,主要侵及肝脏、心肌、肾脏及骨骼肌,同时累及肝脏、骨骼肌、心肌的类型有GSD IIIa型和GSD IV型。由于糖原利用障碍,糖异生增加,糖酵解增加,出现GSD典型临床表现“三高一低”:高脂血症、高乳酸血症、高尿酸血症、空腹酮症性低血糖。异常的糖原在肝脏沉积过多引起明显的肝大,反复低血糖引起发育迟缓及智力低下。
GSD Ⅲ型(OMIM232400)是由位于染色体1p21上的AGL基因突变导致糖原脱支酶(4-α-葡聚糖基转移酶和淀粉-1,6-葡萄糖苷酶)缺陷引起糖原支链不能被分解,导致大量形态结构异常的极限糊精在患者的肝脏和(或)骨骼肌、心肌中堆积[1]。由于AGL基因突变对不同酶活性功能的影响,临床将GSD Ⅲ分为4个亚型。GSD IIIa型(同时累及肝脏和肌肉)占GSD III型病例的85%,GSD IIIb型约占15%(仅累及肝脏),其他两型为4-α-葡聚糖基转移酶和淀粉-1,6-葡萄糖苷酶选择性缺陷,临床比较罕见。
GSD IIIa型临床表现多变,空腹低血糖、高脂血症,肝脏肿大、转氨酶升高,肌无力、肌酸激酶(CK)升高是其特征性表现。然而肝脏症状通常随年龄增长而改善,大部分病例在青春期后恢复正常,部分患者在成人期仅出现肌肉受累,这给成人病例的临床诊断带来困难。Derk团队一项欧洲GSD III型患者的多中心、大样本、回顾性研究总结了不同症状的发生率:肝大95%、肝硬化6.3%,肝腺瘤6.9%、肝癌1.7%、左心室肥厚40.4%、CK增高81%、运动能力下降52.3%、高甘油三酯血症85.4%、高胆固醇62.8%。
非侵入性基因检测是确诊GSD 的主要方法,肝脏组织病理学能够协助不同类型GSD之间进行鉴别。GSD III型经典的肝脏病理改变包括:植物细胞样改变,糖原颗粒被过碘酸-希夫(periodic acid-Schiff, PAS)染色阳性、被淀粉酶消化,同时可有脂肪变性、轻度肝细胞周围纤维化、肝硬化表现[2]。
GSDIII型目前尚无特异性治疗,主要在于改变生活方式,特别是饮食和锻炼。治疗目标为避免低血糖发作,纠正代谢紊乱,减少或延迟严重并发症发生。应避免禁食,幼儿期可以喂养生玉米淀粉(一次0.5-1g/kg,一日4-5次)并夜间加餐来维持正常血糖。注意调整饮食结构,推荐高蛋白(每日3g/kg)、高脂肪、低碳水化合物饮食,避免单糖摄入。运动方面可以适当进行有氧训练和力量训练,在锻炼过程中注意监测血糖水平,锻炼建议需要结合患者的代谢和肌肉病理特点,避免过度运动带来的负面影响。目前基因治疗仍在探索中,已出现严重肝硬化的患者可能最终需要肝移植治疗。
本期月报分享一例成人IIIa型GSD病例,希望能拓展临床医师的诊断思路,当临床遇到不明原因肝肿大,伴肝脏及肌肉损伤标志物升高的患者时注意鉴别此诊断。早期识别,及时干预,帮助患者减少或延迟严重并发症发生。
参考文献:
[1] Hannah WB, Derks TGJ, Drumm ML, Grünert SC, Kishnani PS, Vissing J. Glycogen storage diseases. Nat Rev Dis Primers. 2023 Sep 7;9(1):46.
[2] Kishnani PS, Austin SL, Arn P, Bali DS, Boney A, Case LE, Chung WK, Desai DM, El-Gharbawy A, Haller R, Smit GP, Smith AD, Hobson-Webb LD, Wechsler SB, Weinstein DA, Watson MS, ACMG. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010 Jul;12(7):446-63.

李新华
医学博士、主任医师、博士研究生导师
中山大学附属第三医院感染科副主任(疑难肝病)
中山大学附属第三医院罕见病中心副主任(罕见肝病)
中华医学会肝病分会遗传代谢性肝病协作组委员
广东省医师协会肝病专科医师分会副主任委员
广东省医师协会肝病医师分会疑难肝病专业组副组长
广东省肝脏病学会肝炎专业委员会常委
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.AAV双基因治疗和雷帕霉素的协同作用挽救了糖原贮积症III型的肌肉和肝脏表型(JCI insight, 2024, IF=6.30; Q1区)

2.糖原贮积症III型:一项评估疾病负担的混合方法研究(Therapeutic advances in endocrinology and metabolism, 2024, IF=3.90; Q2区)
3.法国关于糖原贮积症III型的管理建议(European journal of medical research, 2023, IF=2.80; Q2区)
4.体育锻炼和高蛋白饮食改善了庞贝病患儿的肌肉力量、父母的疲劳和身体生活质量(Journal of inherited metabolic disease, 2023, IF=4.20; Q2区)
5.缓释玉米淀粉在肝糖原贮积症患者中的短期和长期可接受性和有效性研究(Orphanet journal of rare diseases, 2024, IF=3.40; Q2区)
6.IV型肝糖原贮积症的自然史研究以及与Gbe 1 ysys模型比较(JCI insight, 2024, IF=6.30; Q1区)
7.综述:肝糖原贮积症内分泌参与病理生理学和护理意义(Reviews in endocrine & metabolic disorders, 2024, IF=6.90; Q1区)
8.综述:α1抗胰蛋白酶缺乏症的罕见变异(Orphanet journal of rare diseases, 2024, IF=3.40; Q2区)
9.综述:遗传性出血性毛细血管扩张症的肝脏表现(Liver international, 2024, IF=6; Q1区)
10.综述:急性肝衰竭的遗传病因(Journal of inherited metabolic disease, 2024, IF=4.20; Q2区)
三、临床资讯
3.1 病例分享:IIIa型糖原贮积症1例
患者男性,19岁,因“反复转氨酶升高18年余”于2023年11月7日入院。
现病史:患者自出生后数月检查发现转氨酶升高(具体不详),未用药。后成长过程中反复多次复查肝功能均有ALT、AST轻度升高(1-2倍正常值上限),无自觉特殊不适,未予处理。入院前1周因体检再次发现转氨酶升高,肝胆彩超提示肝硬化、肝脾轻度增大。遂来我院进一步就诊。
既往史、个人史、家族史:既往体健,流行病学史无特殊;生长发育史同同龄人,未婚未育;家族中无类似患者。
体格检查:神志清楚,对答切题,肝掌(-)、蜘蛛痣(-)、胸壁皮肤毛细血管扩张(-),皮肤巩膜无黄染,球结膜未见水肿,心肺无特殊,腹部平坦,腹壁静脉未见显露,腹软,无压痛、反跳痛,未触及包块,Murphy's征(-),肝脏、脾脏肋下未触及,肝区无叩击痛,移动性浊音阴性,肠鸣音正常。双下肢无水肿。
入院后完善检查:
肝功能:AST 89U/L↑(正常值15-40U/L),ALT 91U/L↑(正常值3-35U/L),GGT 98U/L↑(正常值10-60U/L),ALP138U/L↑(正常值45-125U/L),TBA 5.3μmol/L,CHE 4368U/L;
心肌酶谱:CK 991U/L↑(正常值18-198U/L),CK-MB 29U/L,LDH 326U/L;
血清尿酸:559 umol/L↑(正常值149-416μmol/L);
血常规、空腹血糖、血脂、肾功能、血乳酸均正常;
病毒性肝炎系列:甲乙丙戊病毒性肝炎抗原抗体均阴性;
自身免疫肝病抗体阴性,IgG4抗体:2.59g/L↑(正常值0.08-1.51g/L),甲功正常;
铜蓝蛋白:0.186g/L↓(正常值0.21-0.53g/L),甲胎蛋白:5.19ng/ml,血清铁蛋白:256.7ng/ml↑(正常值15-200ng/mL);
心电图:未见异常;
肝胆脾胰彩超:肝硬化声像,肝增大,肝内异常回声区,考虑肝硬化结节可能性大。门静脉、肝静脉、肠系膜上静脉、脾静脉血流通畅。胆囊超声检查未见明显异常。肝内外胆管未见扩张。胰腺超声检查未见明显异常。脾轻度增大。
上腹部MR平扫+增强+呼吸门控3.0T+磁共振功能成像:1.肝硬化,脾稍大。2.肝内多发不典型增生结节。3.肝内散在异常灌注灶。(图1)
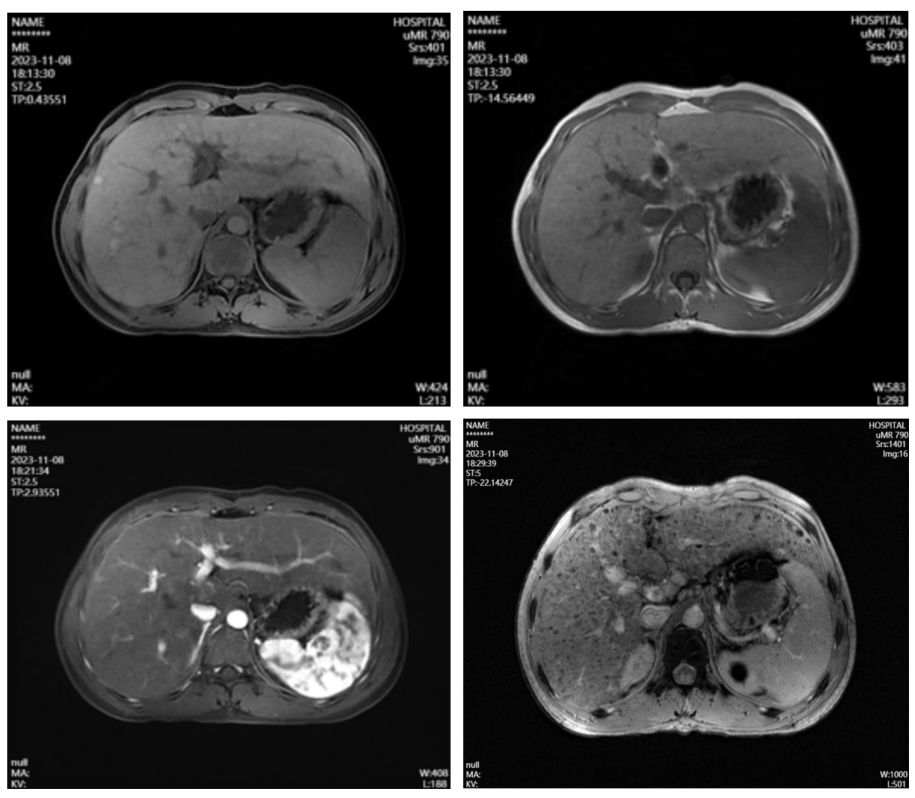
图1.腹部核磁
肝穿病理:(图2)
(肝脏组织)肝小叶结构破坏,肝细胞广泛水肿、部分气球样变,较多肝细胞可见糖原核,可见点状坏死及灶性坏死,肝细胞少量见脂肪变性(约2%),未见胆色素颗粒沉积;门管区扩大,较多淋巴细胞、少量浆细胞、中性粒细胞及嗜酸性粒细胞浸润,可见轻度界板坏死及少量桥接坏死,纤维组织增生,分割肝组织形成假小叶,少量小胆管上皮损伤、变性,形态结合免疫组化、特殊染色,符合慢性肝病(G3S4),考虑遗传代谢性肝病,请结合临床其他检查综合考虑。
免疫组化结果:CK19、CK7(小胆管+),HBcAg(-),HBsAg(-);
特殊染色结果:网状纤维染色、Masson三色染色(示纤维组织增生),D-PAS(示小胆管基底膜不完整),铁染色(-),铜染色(-)。
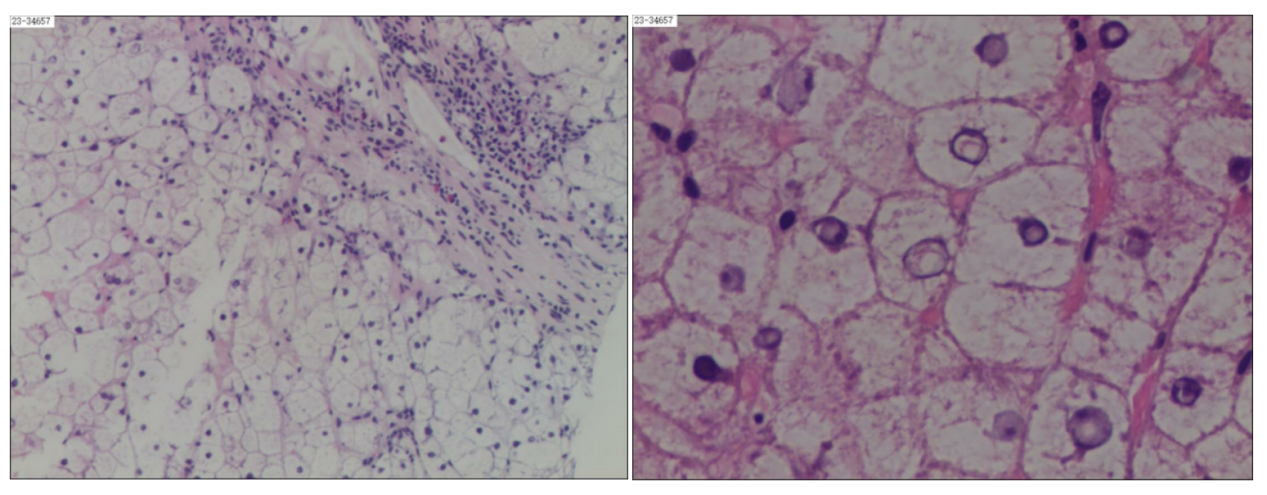
图2.肝脏病理(HE染色,右图箭头所示为糖原核)
全外显子基因测序:AGL基因检测到2个杂合突变:c.2569C>T(p.Gln857Ter)、c.846+4A>G(剪接突变)。(图3)
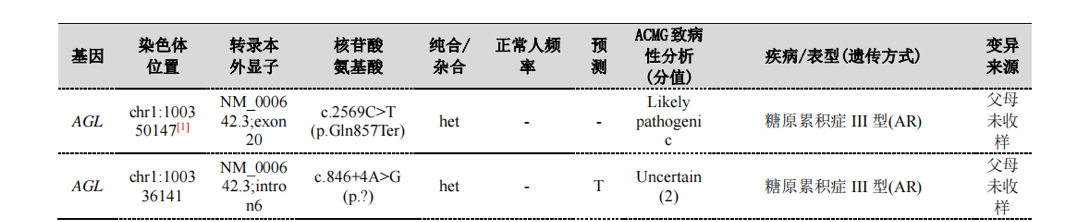
图3.全外显子基因测序结果
诊断经过:患者青年男性,反复肝功能异常,伴有肌酸激酶升高、高尿酸血症,影像学提示肝硬化,肝脏肿大。虽无明显空腹低血糖、高脂血症、高乳酸血症表现,但同时出现肝脏、肌肉损伤,需高度警惕糖原代谢病可能,尤其是GSD IIIa或IV型。遂进一步予患者完善肝穿活检及全外显子基因测序检查:肝脏病理提示较多肝细胞可见糖原核,基因检测证实存在AGL基因2个致病性突变。综上,最终诊断GSD IIIa型。
治疗与随访:诊断明确后给予患者饮食指导:夜间加餐、避免低血糖、避免单糖摄入。调整饮食结构:脂肪/蛋白质/碳水化合物(2:2:1)。给予药物治疗:还原型谷胱甘肽、辅酶Q10。出院后随诊复查肝功能、心肌酶谱逐步恢复正常。
3.2 供稿专家简介
李新华
医学博士、主任医师、博士研究生导师
中山大学附属第三医院感染科副主任(疑难肝病)
中山大学附属第三医院罕见病中心副主任(罕见肝病)
中华医学会肝病分会遗传代谢性肝病协作组委员
广东省医师协会肝病专科医师分会副主任委员
广东省医师协会肝病医师分会疑难肝病专业组副组长
广东省肝脏病学会肝炎专业委员会常委

许镇
医学博士、副主任医师、硕士研究生导师
中山大学附属第三医院感染性疾病科
广东省药学会肝脏病专家委员会秘书
广东省肝脏病学会肝炎专业委员会委员
广东省预防医学会感染病学专业委员会委员
广东省临床医学学会肺癌精准治疗及临床研究专业委员会青年委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间