
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张敏
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
遗传性果糖不耐受症(HFI)是一种罕见的常染色体隐性遗传(AR)疾病,由于9q22.3染色体上的醛缩酶B亚型基因(ALDOB)变异,导致肝脏、肾脏和肠道中醛缩酶B缺乏。特征为摄取果糖、蔗糖或山梨醇后发生严重的低血糖,若不及时终止此类食物,会导致肝肾功能损伤及生长发育障碍。
在HFI患者中的大多数突变仅限于单一的种族群体。HFI患者中ALDOB基因最常见的两种变异是p.(Ala150Pro)和p.(Ala175Asp),约占全世界HFI已发现所有变异等位基因的68%,仅p.(Ala150Pro)变异就占53%,但在不同地理区域之间具有不同频率。在中国人群中p.(Ala150Pro)、p.(Ala175Asp)等位基因频率显著降低,而p.(Ala338Val)、p.(Ala338Gly) 两种变异等位基因频率显著高于非芬兰欧洲人群。中国人HFI患病率较低,估计约为 (1/504,678),远低于非芬兰欧洲人 (1/23,147)、芬兰境内的芬兰人 (1/55,539)、混血美国人 (1/132,801) 和德系犹太人 (1/263,150)[1-3]。
正常人摄入果糖后,生成1-磷酸果糖(F-1P),醛缩酶B催化F-1P裂解为二羟丙酮磷酸(DHAP)和D-甘油醛。果糖不耐受患者醛缩酶基因发生突变,使醛缩酶B活性下降甚至消失,导致肝和肾中F-1P堆积,除导致肝大和直接损害肝肾细胞外,累积的F-1P还导致肝脏中一些其它酶活性受抑,包括磷酸化酶、果糖1,6-二磷酸酶、肝醛缩酶和果糖激酶,结果使肝糖原分解和糖异生都发生障碍而导致低血糖症的发生。例如过多的F-1P抑制了磷酸化酶的作用,阻碍糖原转变成葡萄糖,导致低血糖;在发生低血糖症之前,F-1P堆积使血中无机磷水平降低,磷酸盐减少和肝脏1-磷酸脱胺酶活化,导致ATP降解增加和血循环中尿酸增高;F-1P的累积和ATP供应不足两者也阻碍了糖原释出1-磷酸葡萄糖;ATP消耗会导致镁释放增加,蛋白质合成受损、肝肾功能障碍的超微结构病变;此外,F-1P增加丙酮酸激酶活性激活糖酵解途径,而醛缩酶B功能障碍时无法将DHAP和甘油醛3-磷酸转化为1,6-二磷酸果糖,可发生乳酸性酸中毒[4,5]。
综上,HFI表现多样,除了肝肿大、肝肾功能障碍外,患者还会出现酮症性低血糖、呕吐、恶心、昏迷、高尿酸、乳酸酸中毒等症状。持久的含果糖饮食会造成患儿肝细胞损伤、坏死、脂肪浸润、胆小管增生和纤维化,甚至肝硬化,其机制可能与F-1P的细胞毒性作用或与缺乏ATP相关,具体机制有待进一步阐明。
HFI作为一种罕见病,临床特征变化大,症状轻重不一,部分病人由于自动回避果糖甜食,可以延迟或隐匿发病,容易漏诊,且由于其临床表现部分类似于肝糖原贮积症等其它糖代谢异常疾病,可能出现误诊。确诊HFI的一般依据:1.从食物中摒除果糖后,临床症状迅速消失;2.果糖耐量试验:一次给予果糖200~250mg/kg静脉快速注射后检测血液中果糖、葡萄糖、无机磷、尿酸和转氨酶可供诊断。阳性结果显示血葡萄糖及血磷急速下降同时果糖、脂肪酸及乳酸上升。但本试验易引起低血糖发作故宜慎用;3.活检肝组织、肠黏膜测定醛缩酶活性显著减低;4.基因检测提示ALDOB复合杂合或纯合突变。
HFI目前缺乏有效药物治疗,主要以对症治疗及饮食控制为主。一旦确定诊断应立即终止一切含果糖和蔗糖食物。低血糖发作时,可予静脉注射葡萄糖缓解并纠正电解质紊乱。还有报告用大量叶酸似乎是有希望的治疗方法,但需更多证据[6,7]。本病经过及时治疗,预后良好,无果糖饮食可使患儿完全正常地生活,但多数仍留有轻度肝肿大和脂肪变性。
本期月报报道一例因肝功能异常、肝大求治的1岁患儿,曾出现低血糖反应,伴生长发育落后,临床初步考虑肝糖原贮积症可能,但不完全符合。遂行基因检查,最终诊断为遗传性果糖不耐受症。通过对该病例的复习介绍,期望临床医师认识该病,对于不明原因的肝功异常,同时伴有发育迟缓、回避含糖饮食的患者,需警惕遗传性果糖不耐受症可能。
参考文献:
1. Schrodi SJ, DeBarber A, He M, et al. Prevalence estimation for monogenic autosomal recessive diseases using population-based genetic data. Hum Genet. 2015;134:659-69.
2. Pinheiro FC, Sperb-Ludwig F, Schwartz IVD. Epidemiological aspects of hereditary fructose intolerance: A database study. Hum Mutat. 2021;42:1548-1566.
3. Tang, M, Chen, X, Ni, Q, et al. Estimation of hereditary fructose intolerance prevalence in the Chinese population. Orphanet J Rare Dis. 2022; 17: 326.
4. Bouteldja N, Timson DJ. The biochemical basis of hereditary fructose intolerance. J Inherit Metab Dis. 2010;33:105-12.
5. Hwang JJ, Jiang L, Hamza M, et al. The human brain produces fructose from glucose. JCI Insight. 2017;2:e90508.
6. Di Dato F, Spadarella S, Puoti MG, et al. Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance. Nutrients. 2019;11:2397.
7. Kim MS, Moon JS, Kim MJ, et al. Hereditary Fructose Intolerance Diagnosed in Adulthood. Gut Liver. 2021;15:142-145.

张敏
医学博士,博士生导师
解放军总医院第五医学中心感染病学部主任医师
中华医学会肝病学分会遗传代谢性疾病学组委员
中华医学会医学遗传学分会生化和代谢学组委员
北京医学会遗传代谢病分会委员
全军科学技术委员会神经内科分会专业委员会委员
北京医学会感染病学分会第一届青年委员
从事青少年乙丙型病毒性肝炎、遗传代谢病/疑难肝病等的诊治及科研工作
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.评估饮食干预措施作为果糖不耐受的管理(Journal of pediatric gastroenterology and nutrition, 2024, IF=2.40; Q1区)

2.遗传性果糖不耐受诊断和管理的临床实践指南(Diseases, 2024, IF=2.90; Q2区)
3.吉尔伯特综合征的营养——根据 PRISMA 声明的临床试验系统评价(Nutrients, 2024, IF=4.80; Q1区)
4.治疗后临床稳定的威尔逊病患者非铜蓝蛋白结合铜和尿铜:与推荐目标的一致性研究(JHEP reports : innovation in hepatology, 2024, IF=9.50; Q1区)
5.伊利诺伊州新生儿酸性鞘磷脂酶缺乏症筛查(Journal of inherited metabolic disease, 2024, IF=4.20; Q1区)
6.I型半乳糖血症个体的临床和生化表型、基因型和长期结果(Molecular genetics and metabolism reports, 2024, IF=1.80; Q3区)
7.病例报道:在腹痛患者中诊断出的遗传性果糖不耐受(Journal of clinical medicine, 2024, IF=3.00; Q1区)
8.病例报道:PMM2-CDG和遗传性果糖不耐受综合诊断的轻症患者(Molecular genetics and metabolism, 2023, IF=3.70; Q2区)
9.病例报道:HJV突变引起血色病:一对双胞胎可变表型(Haematologica, 2024, IF=8.20; Q1区)
10.病例报道:三名晚期诊断的经典半乳糖血症患者的自然病程(Molecular genetics and metabolism reports, 2024, IF=1.80; Q3区)
三、临床资讯
3.1 病例分享:一例遗传性果糖不耐受症
患儿男,1岁1月,因“间断发热4天、肝功异常2天”就诊。
现病史:缘于2023年8月17日患儿无明显诱因出现咳嗽,少量白痰,家长带至当地诊所就诊,考虑“上呼吸道感染”应用“头孢氨苄颗粒、小儿化痰止咳颗粒、复方甘草口服溶液”等治疗。8月17日午睡后出现呼之不应,即于当地医院就诊,化验:血糖1.8mmol/L, ALT 759U/L,AST 2327U/L,TBil 9.8μmol/L,DBil 7.4μmol/L,WBC 8.88×109/L,N 24.8%,RBC 4.86×1012/L,HGB 122g/L,PLT 386×109/L。B超提示“肝大伴回声增强”。考虑低血糖反应、上呼吸道感染、肝损害,即予口服高糖2支对症治疗,后神志转清。予以头孢噻肟钠、盐酸氨溴索、复方甘草酸苷等抗感染、保肝对症治疗。为进一步诊治于2023年8月21日来我院急诊就诊,收入我科。
既往史及个人史:生于原籍,第1胎,足月剖宫产,母乳喂养。否认伤寒、结核等传染病史,否认慢性病史,否认手术及外伤史,否认药物过敏史,按时进行疫苗预防接种。
家族史:父母体健,非近亲结婚,否认遗传性疾病史。
入院查体:体温 36.9℃,脉搏 120次/分,呼吸:24次/分,血压:85/47mmHg,身高1800px,体重8.8kg。发育稍落后,营养中等,自动体位,查体合作。神志清,精神可,面色正常,皮肤、巩膜无黄染,肝掌阴性,未见蜘蛛痣。全身浅表淋巴结未扪及肿大。心肺未见异常。腹部饱满。未见腹部静脉曲张,全腹软,无压痛及反跳痛,肝右肋下125px,剑突下75px,莫菲氏征阴性,脾左肋下未及,肝上界位于右锁骨中线第Ⅴ肋间,肝、脾、肾区无叩痛,移动性浊音阴性,肠鸣音正常,双下肢无水肿。生理性反射存在,病理性反射未引出。
入院后完善检查:
血常规:WBC 14.2×109/L,N 10.3%,RBC 5.01×1012/L,HGB 124g/L,PLT 461×109/L。
生化:ALB 48g/L,TBil 6.2µmol/L,DBil 1.6µmol/L,ALT 247U/L,AST 123U/L,ALP 211U/L,GGT 96U/L。肾功能、电解质、尿酸、乳酸、血脂均正常。
甲、乙、丙、戊病毒标记物均阴性。CMV及EBV IgG、IgM、DNA均阴性。铜蓝蛋白0.34g/L。自身抗体系列均阴性。免疫球蛋白水平均正常。淋巴细胞亚群水平正常。血尿质谱检查阴性。
腹部B超:“肝脏增大、肝脏回声增粗、密集,肝右叶最大斜径2925px,肝左叶上下径70mm,前后径1400px。”
诊断分析:
患儿入院前有上呼吸道感染,经对症处理已稳定。主要因肝功异常、肝大来诊。本次发病前曾有服药史,经停用可疑损肝药物及保肝治疗后肝功较前好转,当地考虑药物性肝损害可能,但需除外其他病因。完善检查后可除外病毒性肝炎、自身免疫性肝病、肝豆状核变性、非酒精性脂肪性肝病、梗阻性黄疸等。
该患儿病例特点为肝损害的基础上1. 生长发育略落后,身高体重仅位于同龄幼儿第三个百分位;2.曾出现严重低血糖反应,且饮食正常情况下有空腹血糖水平偏低史; 3. B超提示肝脏增大,脾脏大小正常。以上特点部分符合糖原贮积症,然而患儿无高尿酸、高乳酸、高甘油三酯、中性粒细胞下降等I型糖原贮积症的其它典型表现,主病诊断存疑,需考虑是否存在其它糖代谢异常相关遗传代谢病。因此建议行肝穿病理和/或全外显子基因测序,患者家属拒绝肝穿,同意行基因检查,遂予完善全外显子基因测序。
基因检测结果:1.本次分析未发现与受检者临床表型显著相关的具有可能临床意义的基因变异(SNV和Indel)。2.受检者在chr9:104187883-104189197区域疑似存在纯合缺失变异。建议结合临床进一步分析,同时进行遗传咨询。
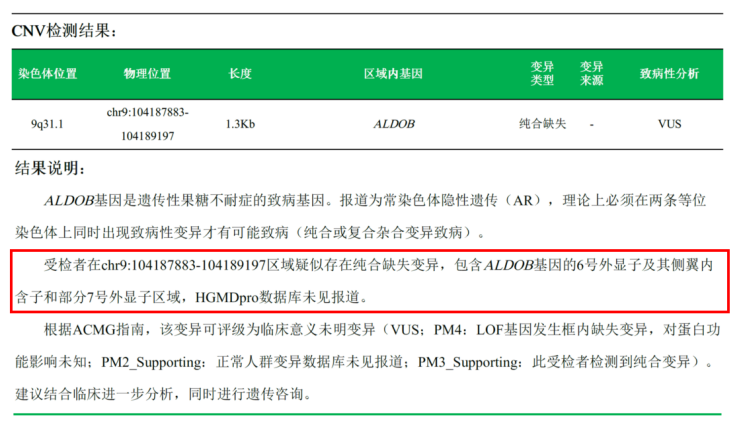
图1. CNV检测结果及说明

图2. 二代测序BAM图
最终诊断:本患儿检出ALDOB基因纯合缺失,虽然ACMG结果为意义未明变异(VUS),但追问病史,患儿昏迷前服用的“头孢氨苄颗粒、小儿化痰止咳颗粒、复方甘草口服溶液” 等味甜,可能含有蔗糖成份,且患者自6个月添加辅食后食用水果会出现呕吐,后有拒食水果及糖果类食品倾向。结合患儿生长发育落后、进食含果糖食物后出现严重低血糖反应、肝脏增大、肝功异常,ALDOB基因纯合缺失,故最终诊断遗传性果糖不耐受症。
治疗转归:本例患儿给予控制蔗糖、果糖饮食摄入及对症治疗。于2024年2月患者再次入院复查:查体身高1875px,体重11kg,化验肝功:ALB 43g/L,TBil 6.5µmol/L,DBil 1.9µmol/L,ALT 63U/L,AST 74U/L,ALP 257U/L,GGT 30U/L,腹部B超提示“轻中度脂肪肝。肝右叶最大斜径2450px,肝左叶上下径67mm,前后径1250px”。肝脏较前缩小。肝脏瞬时弹性检查肝脏弹性5.1Kpa、CAP 290db/m。嘱患儿家长继续严格饮食控制,避免果糖蔗糖类摄入,临床随访观察,建议定期评估复查。
3.2 供稿专家简介
张敏
医学博士,博士生导师
解放军总医院第五医学中心感染病学部主任医师
中华医学会肝病学分会遗传代谢性疾病学组委员
中华医学会医学遗传学分会生化和代谢学组委员
北京医学会遗传代谢病分会委员
全军科学技术委员会神经内科分会专业委员会委员
北京医学会感染病学分会第一届青年委员
从事青少年乙丙型病毒性肝炎、遗传代谢病/疑难肝病等的诊治及科研工作

冯丹妮
医学硕士
解放军总医院第五医学中心肝病医学部 主治医师
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间