
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会;中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:侯维
执行编辑:郑素军,於海天,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
2025年2月28日是第18个国际罕见病日,今年的主题是“不止罕见”(More than you can imagine),旨在强调社会不应只关注患者的罕见病本身,更要看到罕见病患者展现出的生命力与可能性。同时罕见病不应是少数人关心的问题,而需要全社会共同关注并应对挑战。在这个特殊的日子中,“遗传代谢性肝病月报”邀请首都医科大学附属北京佑安医院侯维教授为大家献上一例跨越8年才获得明确诊断的罕见病例。

一、主编致辞
端粒是人类染色体末端的重复DNA序列,其与一组特殊的保护蛋白(由6种蛋白组成Shelterin复合体)结合,足够的端粒长度与完整的保护蛋白结构有助于保护染色体末端免于降解、维持基因组完整性[1]。生理状态下端粒会随年龄增长而缩短,过度端粒缩短则预示着早衰的发生[2]。短端粒综合症(Short Telomere Syndrome, STS)是负责维持和修复端粒的基因变异导致端粒长度过度缩短的一组孟德尔遗传性疾病。短端粒综合征致病基因种类繁多,包括编码端粒蛋白复合体的基因(TERF1、TERF2、TIN2、RPA1、POT1、ACD)、编码端粒酶及相关因子的基因(TERT、TERC、DKC1、NHP2、NOP10、WRAP53、NAF1、PARN、RTEL1、CTC1、STN1),不同基因变异对应着不同遗传方式。伴随端粒异常缩短出现的退行性并发症是短端粒综合症患者发病和死亡的主要原因。
短端粒综合症导致的退行性并发症,受累器官包括肺、肝、骨髓和皮肤等,端粒缩短程度影响发病年龄和临床表型(图1)。短端粒综合症患者肺间质受累最常见,是端粒轻度缩短的成人患者的首发表现,可表现为特发性肺纤维化和肺气肿,研究发现约30%的家族性肺纤维患者中存在端粒及端粒酶基因突变[3-5]。骨髓衰竭通常是端粒严重缩短的儿童患者的首发表现,骨髓衰竭可以表现为再生障碍性贫血、骨髓增生异常综合征、急性髓性白血病。在罕见的情况下,短端粒综合症会导致先天性角化不全,患者可出现典型的网格状皮肤色素沉着、指/趾甲角化不良和口腔黏膜白斑等特征性临床“ 三联征” 表现[6,7]。短端粒综合症患者还可以表现出其他早衰特征,包括早期白发和骨质疏松,恶性肿瘤易感性增加。
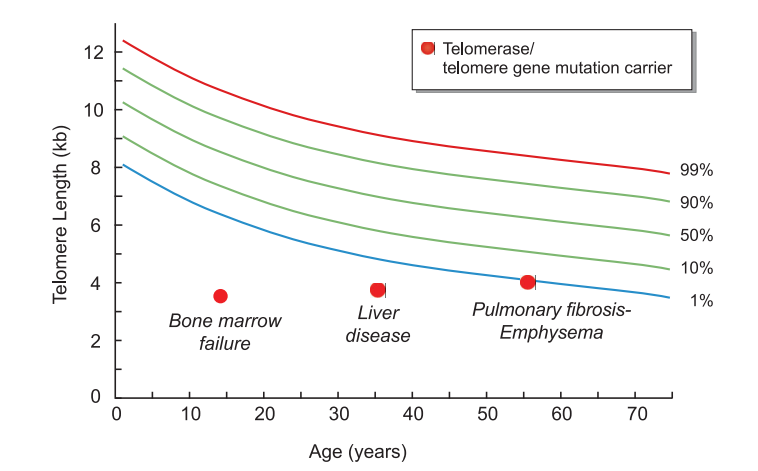
图1. 短端粒综合征端粒长度与发病年龄和临床表型的关系[1]
约10%的短端粒综合症患者出现肝脏并发症[6,8],包括肝纤维化、非硬化门脉高压、肝肺综合征等。其自然病程和病理生理尚未完全了解,早期可表现为脾大、杵状指,后期可出现缺氧、呼吸困难,终末期肝病表现。影像学上可表现为脾大、尾叶肥大和结节性肝脏外观、腹水、静脉曲张表现。肝脏病理主要表现为非肝硬化性门脉高压改变,出现结节性再生性增生,肝脏内外的血管畸形等[9](图2)。基于约翰霍普金斯大学短端粒综合征登记处的数据的研究,共纳入150名端粒综合征的患者,其中42 名患者因呼吸困难(呼吸急促)就诊,研究主要关注无明显肺部疾病(如肺纤维化)但仍然表现出低氧血症的患者。发现在端粒综合征患者中,肝肺综合征的发生率为 21%(9/42)。这些患者的平均发病年龄为25岁,远早于肺纤维化的典型发病年龄(55岁)[9]。
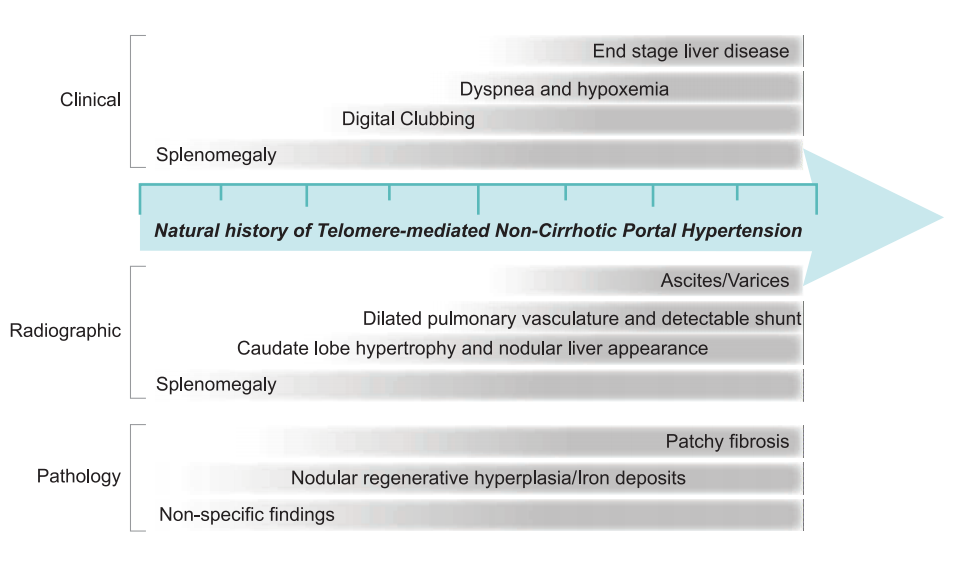
图2. 短端粒综合征导致非硬化性门脉高压的疾病自然史和影像学、病理学表现[3]
研究推测,端粒功能障碍可能通过以下机制导致非肝硬化性门脉高压和肝肺综合征[3]。①肝脏微血管功能异常。端粒缩短可能导致肝内皮细胞衰老和功能障碍,导致血管重塑和动静脉畸形。血管异常可能引发肝内血流动力学变化,最终导致门静脉高压和肺内血管扩张,形成肝肺综合征。②肝组织再生障碍。端粒综合征患者常见结节性再生性增生,但没有典型的肝硬化表现。可能的机制包括慢性微血管缺血和局部肝细胞增生代偿,导致肝脏形态变化。③铁代谢异常。研究发现部分患者肝组织存在非输血相关的铁沉积。可能与慢性血管损伤和红细胞代谢异常有关,进一步加重了肝脏病理变化。
器官移植(骨髓、肺、肝移植)仍然是治疗与短端粒综合征相关器官衰竭的主要手段。无法接受骨髓移植的患者可以尝试雄激素治疗,据报道血液学应答率约为50%。体内外实验证实雄激素通过上调端粒酶基因表达,从而减缓端粒磨损速率并增强细胞再生。但治疗期间需监测转氨酶升高、血脂异常、生长加速、雄激素效应等不良反应[10]。
本期月报报道一例20岁的短端粒综合征患者,以骨髓衰竭和非硬化性门脉高压为主要临床表现。患者血白细胞和血小板呈进行性下降趋势,骨髓穿刺未见巨核细胞,血小板减少,提示血小板生成障碍。肝穿刺病理及腹部核磁结果符合非硬化性门脉高压。基因检测存在TERT基因杂合突变c.336delC(致病性变异),既往有文献报道该致病位点可表现为端粒相关的骨髓造血功能衰竭和/或肺间质纤维化[11]。本例患者尚无呼吸困难、低氧血症等呼吸系统表现,肺部CT亦无明显肺纤维化,提示短端粒综合征临床表型、发病年龄和严重程度与端粒的长度相关。本期月报通过分享该病例为短端粒综合症患者的肝病表型提供了参考,有助于我们拓展该病的诊断思路,加深认识,缩短诊断时间。
参考文献:
[1] Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012; 13 ( 10 ): 693-704.
[2] Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts . Nature. 1990; 345 (6274): 458-460.
[3] Alder JK, Stanley SE, Wagner CL, et al. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015; 147 ( 5 ): 1361-1368.
[4]Cogan JD, Kropski JA, Zhao M, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015; 191 (6): 646-655.
[5]Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015; 47 (5): 512-517 .
[6]Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000; 110 (4): 768-779.
[7] Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. 2009; 23 (2): 215-231.
[8] Calado RTRJ, Regal JA, Kleiner DE, et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS One. 2009; 4 (11): e7926.
[9] Gorgy AI, Jonassaint NL, Stanley SE, et al. Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. Chest. 2015 Oct;148(4):1019-1026.
[10] Mangaonkar AA, Patnaik MM. Short Telomere Syndromes in Clinical Practice: Bridging Bench and Bedside. Mayo Clin Proc. 2018 Jul;93(7):904-916.
[11] Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007 Mar 29;356(13):1317-26.

侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.端粒生物学障碍患者的肝脏疾病和移植:一项国际多中心队列研究(Hepatology communications, 2024, IF=5.60; Q1区)

2.特发性门窦血管疾病中短端粒综合征的高患病率(Hepatology communications,2024, IF=5.60; Q1区)
3.晚发性端粒生物学障碍:来自回顾性登记队列的临床见解和治疗结果(Blood advances,2025, IF=7.40; Q1区)
4.年轻的HBV相关HCC中男性具有TERT基因变异和HBV整合优势:性别差异影响(Clinical and molecular hepatology,2025, IF=14.00; Q1区)
5.端粒长度检测对间质性肺疾病的临床意义(Chest,2024, IF=9.50; Q1区)
6.端粒生物学障碍中的RTEL1变异(American journal of medical genetics,2025, IF=1.70; Q3区)
7.综述:端粒生物学障碍:聚焦胃肠道和肝脏表现(Current hematologic malignancy reports,2024, IF=2.70; Q2区)
8.综述:端粒生物学障碍患者的肺、肝和异基因造血干细胞移植(Current hematologic malignancy reports,2024, IF=2.70; Q2区)
9.病例报道:TERT基因新发突变相关先天性角化不良伴门窦血管病1例(Journal of medical case reports,2025, IF=0.90; Q3区)
10.病例报道:1例TERT突变相关端粒生物学障碍患者的网状色素改变和Terry指甲(Pediatric dermatology,2025, IF=1.20; Q3区)
三、临床资讯
3.1 病例分享:一例短端粒综合征患者
患者女性,20岁。因“发现肝功异常8年余”于2023年就诊。
现病史:
8年前(2015年)患者家属发现患者“面色暗、眼圈黑”就诊于当地医院,肝功能提示转氨酶、胆红素均异常(具体不详),未规律诊疗。
7年前(2016年)患者就诊于当地医院,化验血常规WBC 4.4×109/L,HGB 140g/L,PLT 120×109/L;肝功能:ALT 63U/L,AST 49U/L,GGT 130U/L,ALP 827U/L,TBIL 23.5μmol/L,DBIL 9.5μmo//L;铜蓝蛋白 0.22g/L;腹部增强CT:脾大,脾静脉稍粗,肝脏右缘欠规则。给予葡醛内酯治疗,此后监测转氨酶、胆红素轻度异常,波动范围基本同前,ALP 降至300U/L,GGT大致同前。期间监测血小板逐渐减低。
6年前(2017年)患者于北京行代谢性肝病panel基因检测,报告提示ATP7B基因复合杂合突变(具体不详)。就诊于北京某医院,查血清铜1.76mg/L,24小时尿铜42.4μg,铜蓝蛋白 18.7mg/dl,眼科会诊未见 KF环,头颅核磁正常。考虑肝豆状核变性不能除外,分别于2017年3月、5月开始接受硫酸锌、青霉铵治疗。2017年5月复查血常规:WBC 3.86×109/L,NEUT% 56.7%,HGB 149g/L,PLT 86×109/L;肝功能:ALT 80U/L,AST 47U/L,GGT 153U/L,ALP 344U/L,TBIL 23.5μmol/L,DBIL 9.5μmol/L。Coombs 试验阴性,尿/便常规、性激素、皮质醇、甲功、ANA/ENA 均正常。2017年8月复查血清铜0.78mg/L,24小时尿铜30.7ug,铜蓝蛋白0.244g/L,EBV/CMV IgM及病毒载量、自免肝5项均正常,腹部超声提示肝实质回声增粗,脾稍大,考虑临床及化验不支持肝豆状核变性诊断,停用硫酸锌及青霉铵,予双环醇25mg tid、熊去氧胆酸250mg bid治疗。
5年前(2018年2月)患者复查血常规大致同前,转氨酶及胆管酶均正常,TBIL 22.8μmol/L,DBIL 7.3μmol/L。2018年8月曾服用中药汤剂1月(具体不详)。2018年11月患者复查血小板逐渐减低至69×109/L,转氨酶及胆管酶均正常,TBIL 32.5μmoI/L。MRCP示肝右叶边缘形态不整伴T2信号不均匀稍高,脾大。行肝穿刺病理:肝组织呈慢性炎,局部肝索拥挤,汇管区未见明确淋巴浆细胞浸润,未见明确细胞坏死,未见明确纤维增生。骨穿提示未见巨核细胞,血小板减少。治疗调整为葡醛内酯联合熊去氧胆酸250mg tid。此后患者未规律诊治。
2年前患者停用保肝药至今,间断复查仍提示肝功能异常,1年前行肝弹性监测为7.2EkPa。入院前(2023-09-11)我院门诊化验示WBC 4.2×109/L,HGB 135g/L,PLT 77×109/L,ALT 60U/L,AST 55U/L,GGT 139U/L,ALP167U/L,TBIL 38.8μmol/L, DBIL 10.5μmol/L。现为进一步明确诊断收入我院。
既往史、个人史、家族史:否认其他既往病史。否认吸烟史,否认饮酒史。无家族类似疾病史。
入院查体:皮肤、巩膜无明显黄染。心、肺听诊无异常,腹部查体无明显异常。
入院后检查:
血常规:WBC 2.9×109/L,HGB 120g/L,PLT 54×109/L;
肝功能:ALT 32U/L,AST 25U/L,GGT 97U/L,ALP 96U/L,TBIL 16μmol/L,DBIL 6μmol/L,ALB 39g/L。
其他化验:凝血功能、甲状腺功能、尿便常规均正常。
病因学:甲肝、乙肝、丙肝、戊肝病毒学标志物阴性,CMV DNA、EBV DNA阴性;lgG 13.9g/L、自身免疫抗体ANA1:100 余均阴性;铜蓝蛋白 0.203 g/L,24h尿铜 31.5ug/L,眼科检査:未见 K-F环,颅脑磁共振成像未见明显异常。
上腹部磁共振增强成像(薄层+动态增强):肝硬化改变,脾大,侧支循环形成。
电子胃镜:慢性非萎缩性胃炎。
肺部CT:右肺下叶微小结节,余无异常。
肝脏病理:
(2018年患者外院肝穿标本送至我院病理科会诊)诊断:肝内慢性淤胆(CK7染色阳性肝细胞及铜沉积),考虑胆源性肝病,请结合临床及基因检测结果分析。
(2023.10.31我院肝穿病理)诊断:(肝穿)汇管区纤维化,门静脉闭塞病,符合特发性非硬化性门脉高压症,伴肝脏结构异常(动脉壁增厚、小胆管增生)。
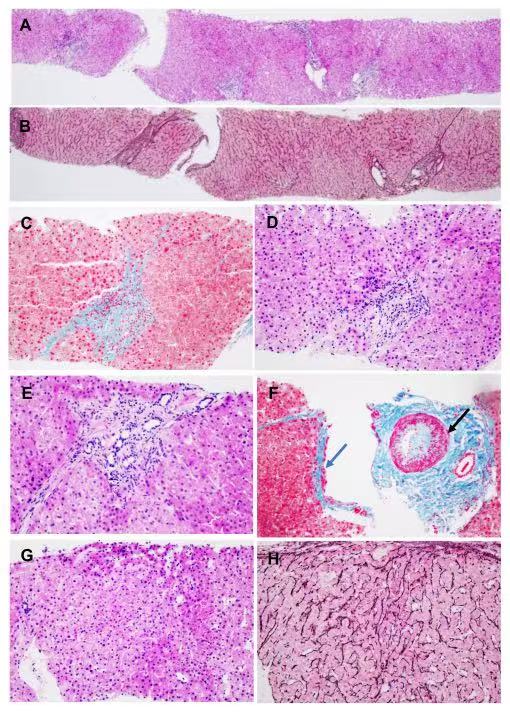
图3.患者2023年肝穿病理。A图(HE染色)和B图(网状纤维染色)未见肝硬化;C图(Masson染色)和D图(HE染色)汇管区间质纤维化伴细隔形成,未见相应口径的门脉分支;E图(HE染色)有的汇管区内见小胆管增生、数目增多;F图(Masson染色)稍大的汇管区内门脉管壁增厚(蓝箭)及动脉壁显著增厚(黑箭);G图(HE染色)和H图(网状纤维染色)肝窦轻度不规则扩张,局部肝板缺血性萎缩。
外周血全外显子基因测序:TERT基因c.336delC (p.P112fs),杂合变异,ACMG评级为致病(图4)。

图4.患者全外显子基因测序结果
端粒长度检测:该样本端粒长度分布为3.58-12.91kb。样本中端粒平均长度为6.19kb,在此年龄段人群中,端粒长度偏短,不符合生物学年龄分布情况(图5)。
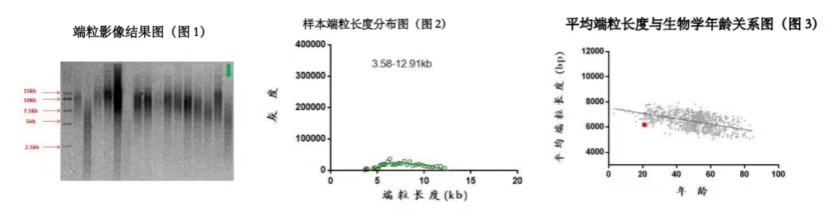
图5.患者端粒长度检测结果
病例特点及诊断分析:
患者青年女性,慢性病程。临床表现为转氨酶及胆管酶轻度升高,伴白细胞及血小板进行性减少。骨髓穿刺提示血小板生成障碍;肝脏核磁提示脾大、侧支循环形成;肝穿发现肝组织内小叶间动脉管壁增厚,小叶间静脉扩张,小叶间胆管数量增多;基因检测存在TERT致病性杂合变异c.336delC;端粒长度检测示端粒长度偏短,不符合生物学年龄分布情况。
综上,患者存在骨髓衰竭及非硬化性门脉高压表现,结合基因检测及端粒长度测定结果诊断为短端粒综合征。
3.2 供稿专家简介
侯维
首都医科大学附属北京佑安医院,肝病中心一科,主任医师
中华医学会感染病学分会青年委员
北京感染病学分会委员
北京医学会肝病学分会青年委员

王晓晓
首都医科大学附属北京佑安医院,肝病中心一科,主治医师,医学博士
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-63291007
▶联系地址:北京市丰台区右安门外西头条8号佑安医院D楼(南入口)405房间