
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:朱磊
执行编辑:郑素军,张思琪,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
一、主编致辞
吉尔伯特综合征(Gilbert Syndrome, GS)与遗传性球形细胞增多症(Hereditary Spherocytosis, HS)分别是胆红素代谢异常和遗传性溶血性贫血中最常见的遗传性疾病[1][2]。GS由编码关键酶尿苷二磷酸葡萄糖醛酸转移酶(UGT1A1)的基因缺陷所致,表现为轻度非结合胆红素升高(17-70 μmol/L),不伴有肝细胞损伤或溶血[3]。大多数GS患者可出现反复性、短暂性黄疸,但约30%的患者无明显症状[3]。而HS是一种因红细胞膜结构蛋白缺陷导致的遗传性溶血性贫血,其特征形态为球形红细胞,此类细胞变形能力差而在脾脏中滞留并破坏[4]。HS的临床表现异质性极高,轻者可无症状,重者可在儿童期即出现需输血干预的严重贫血[4]。GS和HS的共患病率可能高于既往认识,当前估算约为每10万人中15至35例[5]。但其在遗传与分子水平的协同致病机制尚未阐明。
当GS与其他遗传或获得因素共存时,其临床表现可能变得复杂且严重。近年来,随着基因检测的广泛应用,GS与其他遗传性疾病的共存日益被认识到,由此产生的协同致病效应及相关诊断挑战正受到越来越多的临床关注。GS通常遵循常染色体隐性遗传模式[3]。本期报道的病例基因检测显示,患者携带UGT1A1变异c.-41_-40dup(UGT1A1*28)和c.-3275T>G(UGT1A1*60),均来源于母亲,而父亲UGT1A1基因型为野生型。UGT1A1*28纯合变异可使UGTIA1酶活性降低约70%[6],而杂合变异仅降低10%~35%。UGT1A1*60作为表型修饰因子,当与UGT1A1*28 或 UGT1A1*6基因共存时,会进一步削弱酶的功能[7-8]。尽管如此,由于患者携带的UGT1A1变异为顺式杂合(均遗传自母亲),不符合GS遗传模式,且既往研究显示上述两个多态性变异共存时UGT1A1酶活性降低程度尚不足以诊断GS[9-10]。然而,在合并HS所致慢性溶血的情况下,患者所携带的UGT1A1变异很可能是导致其胆红素水平进一步升高及胆石症风险增加的重要因素[11]。
HS主要呈常染色体显性遗传,少数为隐性模式[12]。本例患者基因检测结果显示,其携带父系来源的SLC4A1基因杂合移码变异c.221del(p.Arg74AsnfsTer35)。该变异导致第74位精氨酸的编码序列发生改变,并于下游第35位残基处引入提前终止密码子,最终形成截短蛋白。该精氨酸位于SLC4A1基因编码的Band 3蛋白的N端胞质结构域,此区域对于蛋白与细胞骨架的连接至关重要。因此,该突变会破坏Band 3蛋白与细胞骨架间的相互作用,进而导致蛋白定位异常和功能丧失[13]。然而,发生在Band 3蛋白C端跨膜结构域的突变,常引起蛋白严重错误折叠、完全缺失,并导致更严重的表型(如重度贫血和远端肾小管酸中毒)[14-15]。因此,本例患者的N端突变表现为“低效等位基因”效应,仅诱发轻度溶血,继而刺激骨髓红细胞生成代偿性增强,表现为网织红细胞增多。正是依赖这一高效代偿机制,患者血红蛋白水平维持在接近正常范围,并未出现明显的临床贫血表现。
本病例揭示了UGT1A1变异与HS协同致病的潜在“双重打击”机制,在慢性溶血导致胆红素生成持续过量的背景下,即使残留单个正常的UGT1A1等位基因,其酶活性也可能无法完全代偿,导致对胆红素处理下降、血清胆红素水平升高,HS临床表型更重。这一发现对临床实践具有重要启示,在疑难肝病诊治中,诊断的“一元论”原则可能存在局限。当患者的临床表现不典型、或其严重程度无法用单一疾病合理解释时,临床医生应积极考虑遗传性共病的可能,并积极借助基因检测进行鉴别与确认。这为推进此类疾病的精准诊断与管理提供了重要的思路与依据。
参考文献
1.Goluch Z, Wierzbicka-Rucinska A, Ksiazek E. Nutrition in Gilbert's Syndrome-A Systematic Review of Clinical Trials According to the PRISMA Statement. Nutrients. 2024;16(14). doi:10.3390/nu16142247
2. Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet (London, England). 2008;372(9647):1411-26. doi:10.1016/s0140-6736(08)61588-3
3. Levitan BN, Skvortsov VV, Morozov AV, Serdyukova TV. Modern view on pathogenesis, diagnosis and treatment of Gilbert's syndrome. Medical alphabet. 2023(35):19-22. doi:10.33667/2078-5631-2022-35-19-22
4. Manciu S, Matei E, Trandafir B. Hereditary Spherocytosis - Diagnosis, Surgical Treatment and Outcomes. A Literature Review. Chirurgia. 2017;112(2):110. doi:10.21614/chirurgia.112.2.110
5. Ahel IB, Dekanić KB, Palčevski G, Roganović J. 289 Hereditary spherocytosis associated with Gilbert syndrome – a case report. Abstracts. 2021. doi:10.1136/archdischild-2021-europaediatrics.289
6. Bosma PJ, Chowdhury JR, Bakker C, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. The New England journal of medicine. 1995;333(18):1171-5. doi:10.1056/nejm199511023331802
7. Huang Y-Y, Huang M-J, Yang S-S, Teng H-C, Huang C-S. Variations in the UDP-Glucuronosyltransferase 1A1 Gene for the Development of Unconjugated Hyperbilirubinemia in Taiwanese. Pharmacogenomics. 2008;9(9):1229-35. doi:10.2217/14622416.9.9.1229
8. Tomerak RH, Helal NF, Shaker OG, Yousef MA. Association between the Specific UGT1A1 Promoter Sequence Variant (c-3279T>G) and Unconjugated Neonatal Hyperbilirubinemia. Journal of Tropical Pediatrics. 2016. doi:10.1093/tropej/fmw031
9.Innocenti, F., Grimsley, C., Das, S., Ramírez, J., Cheng, C., Kuttab-Boulos, H., Ratain, M. J., & Di Rienzo, A. (2002). Haplotype structure of the UDP-glucuronosyltransferase 1A1 promoter in different ethnic groups. Pharmacogenetics, 12(9), 725–733. https://doi.org/10.1097/00008571-200212000-00006
10. Maruo, Y., D'Addario, C., Mori, A., Iwai, M., Takahashi, H., Sato, H., & Takeuchi, Y. (2004). Two linked polymorphic mutations (A(TA)7TAA and T-3279G) of UGT1A1 as the principal cause of Gilbert syndrome. Human genetics, 115(6), 525–526. https://doi.org/10.1007/s00439-004-1183-x
11. del Giudice EM, Perrotta S, Nobili B, Specchia C, d'Urzo G, Iolascon A. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood. 1999 Oct 1;94(7):2259-62. pii: S0006-4971(20)71162-6.
12.Trivedi S, Chaudhary SR, Jain P, Agarwal S, Garg V, Singhal U. Twin cases of hereditary spherocytosis: A tale of missed diagnoses. Panacea Journal of Medical Sciences. 2025;15(1):225-30. doi:10.18231/pjms.v.15.i.1.225-230
13.Satchwell TJ, Hawley BR, Bell AJ, Ribeiro ML, Toye AM. The cytoskeletal binding domain of band 3 is required for multiprotein complex formation and retention during erythropoiesis. Haematologica. 2015;100(1):133-42. doi:10.3324/haematol.2014.114538
14.Seon Hee C, Philip S L. Identification of a critical ankyrin-binding loop on the cytoplasmic domain of erythrocyte membrane band 3 by crystal structure analysis and site-directed mutagenesis. J Biol Chem. 2002.
15.Timothy J S, Bethan R H, Amanda J B, M Leticia R, Ashley M T. The cytoskeletal binding domain of band 3 is required for multiprotein complex formation and retention during erythropoiesis. Haematologica. 2014.

朱磊
大连医科大学附属第一医院消化内科主任医师、研究生导师
辽宁省基层卫生协会肝病防治专委会副主任委员
辽宁省医学会肝病学会委员
辽宁省免疫学会肝病与免疫分会常委
辽宁省中西医结合学会消化分会委员
大连市中西医结合学会肝病学分会常委、胆胰内镜学会委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.病例报告:在一例中国吉尔伯特综合征患者中发现新型致病性UGT1A1基因突变(frontiers in medicine,2025,IF=3.0;Q1区)

2.PON-1与UGT1A1基因的单核苷酸多态性变异对重型地中海贫血患者铁螯合治疗结局及锌营养状况的影响(Biological trace element research,2025,IF=3.6;Q2区)
3.化疗诱发非结合性高胆红素血症合并其他触发因素:一例伴有UGT1A1突变相关性吉尔伯特综合征的儿童T细胞急性淋巴细胞白血病病例(Current oncology,2025,IF=3.2;Q2区)
4.循环胆红素水平(而非其遗传决定因素)与青少年脂肪性肝病呈负相关(International journal of molecular sciences,2025,IF=4.9;Q1区)
5.吉尔伯特综合征捐献者的肝脏移植获得成功(Journal of clinical and experimental hepatology,2025,IF=3.2;Q2区)
6.根据液相色谱-质谱联用分析,胆红素代谢不影响血清胆汁酸谱:一项人类病例对照研究(International journal of molecular sciences,2025,IF=4.9;Q1区)
7.红细胞生成性原卟啉症的病理学诊断挑战:病例报告一例(Frontiers in medicine,2025,IF=3.0;Q1区)
8.胆红素代谢在健康与疾病状态下的生理学基础模型构建及其与真实世界数据的比较分析(CPT: pharmacometrics & systems pharmacology,2026,F=3.0;Q2区)
9.因失代偿期肝硬化住院时的高胆红素血症可预测院内感染:来自ATTIRE试验的数据(Hepatology communications,2025,IF=4.6;Q1区)
10.吉尔伯特综合征在英国初级诊疗人群中的患病率及其前驱症状与特征的数据驱动探索研究(Clinical epidemiology,2025,IF=3.2;Q1区)
三、临床资讯
3.1 病例分享:吉尔伯特综合征合并遗传性球形红细胞增多症的“双重打击”:基因诊断揭示高胆红素血症的协同机制
患者男性,39岁,于2025年5月18日因“间断皮肤及巩膜黄染10年,右上腹疼痛2个月”入院。
现病史:10年前患者间断皮肤及巩膜黄染就诊于外院,行肝穿提示肝脏铁沉积及脾肿大,但诊断过程不完整,未能明确病因。近2月来,患者间断性右上腹疼痛,于外院查腹部超声示胆囊多发结石,胆囊沉积物,脾大;肝功检查结果:天冬氨酸氨基转移酶63 IU/L,丙氨酸氨基转移酶210 IU/L,碱性磷酸酶103 U/L,γ谷氨酰转肽酶283 U/L,总胆红素80.8 μmol/L,直接胆红素10 μmol/L。为求进一步诊治收入院。
入院后查体及检查:体温36.8℃,心率68次/分,血压110/73mmHg,身高181 cm,体重65 kg;皮肤及巩膜黄染,无肝掌及蜘蛛痣。腹部平软,右上腹轻压痛,无反跳痛及肌紧张,双下肢不肿。实验室检查:血常规:白细胞7.86×10⁹/L,血小板163×10⁹/L,血红蛋白131 g/L,平均红细胞体积93.8 fL,平均血红蛋白浓度365 g/L,网织红细胞计数7.18%。凝血功能检查:凝血酶原时间11.2秒,凝血酶时间18.3秒,国际标准化比值(INR)0.97。其他结果:甲型、乙型、丙型、戊型肝炎病毒阴性、EB病毒及巨细胞病毒阴性。肝病相关抗体、ANCA、免疫球蛋白(IgG、IgA、IgM)、抗核抗体及滴度、免疫球蛋白G亚型4均为正常。虽然血清铜蓝蛋白水平偏低0.169 g/L(正常参考值范围0.2-0.6g/L),但血清铜及24小时尿铜定量正常,且眼底检查未见KF环,因此Wilson病被排除。
病情分析及诊疗经过:患者临床表现为以非结合胆红素升高为主的慢性高胆红素血症,并伴有脾大及胆结石。这一复杂的临床表现既提示了存在先天性胆红素代谢障碍与溶血过程的可能。该怀疑得到了溶血相关的实验室指标的支持:网织红细胞比例显著增高(7.18%)、外周血涂片可见约11%球形红细胞(图1)、红细胞渗透脆性增加(开始溶血0.6%,完全溶血0.4%),而直接与间接Coombs试验均为阴性,共同指向代偿性慢性溶血,并高度提示遗传性球形红细胞增多症(HS)的可能。为从分子层面明确这一双重病因,我们进行了全外显子组测序(WES)。分析结果清晰地揭示了“双重打击”的遗传学基础。首先,在SLC4A1基因上鉴定出一个杂合移码突变c.221del(p.Arg74AsnfsTer35),家系检测证实其来源于患者父亲,母亲该位点无变异。根据ACMG指南,该变异判断为疑似致病性变异(Likely pathogenic)PVS1+PM2_Supporting,从而确诊HS。此外,测序数据在UGT1A1基因上发现2个杂合变异c.-41-40dup与c.-3275T>G,家系检测证实均来源于患者母亲,为顺式杂合变异。
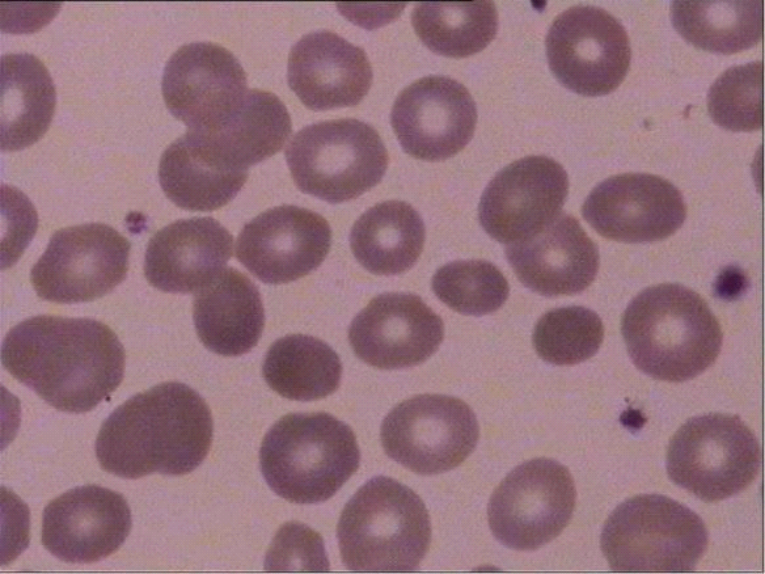
图1 外周血涂片显示小球形细胞
追问病史,患者父母均无胆红素升高病史,为进一步解析SLC4A1基因的致病性,我们开展了整合性的遗传学与生物信息学分析。家系分析证实患者携带父系来源的SLC4A1基因杂合移码突变c.221del(p.Arg74AsnfsTer35)(图2)。
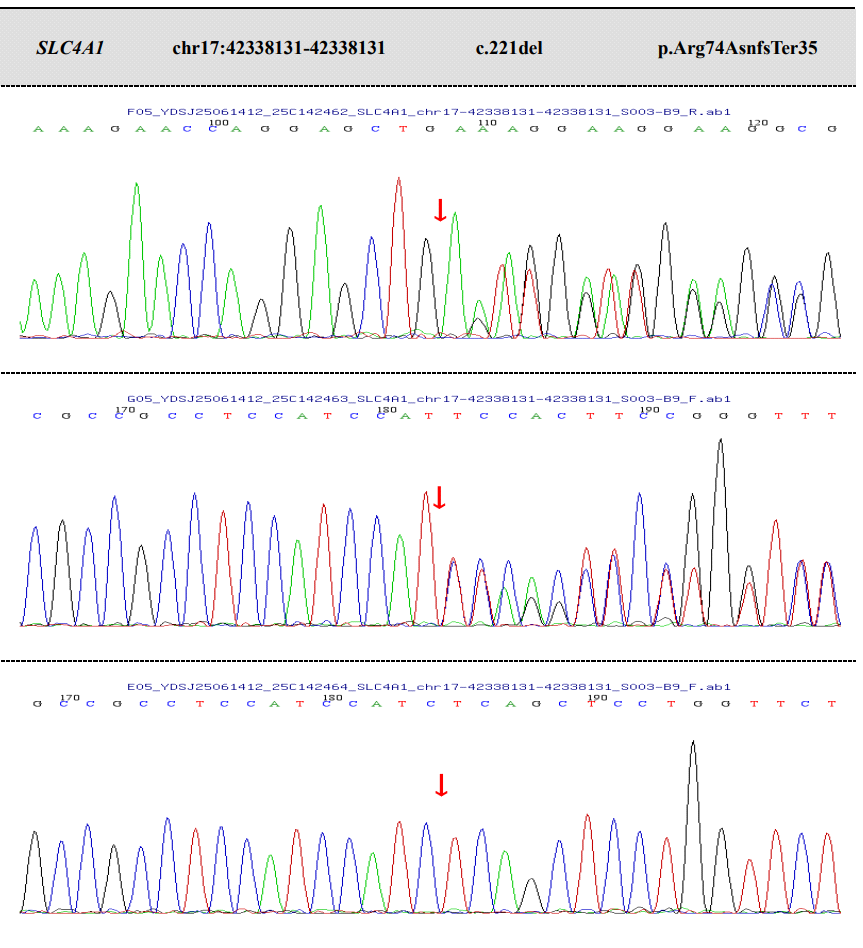
图2 SLC4A1基因c.221del (p.Arg74AsnfsTer35) 变异家系Sanger测序分析
测序图谱显示,在SLC4A1基因 c.221del 位点(箭头所示),先证者(上图)与其父亲(中图)均表现为从该位点开始的序列重叠信号(套峰),符合杂合缺失变异导致的移码特征;其母亲(下图)在该位点为野生型单峰。该结果证实此 c.221del (p.Arg74AsnfsTer35) 移码变异来源于父系遗传。
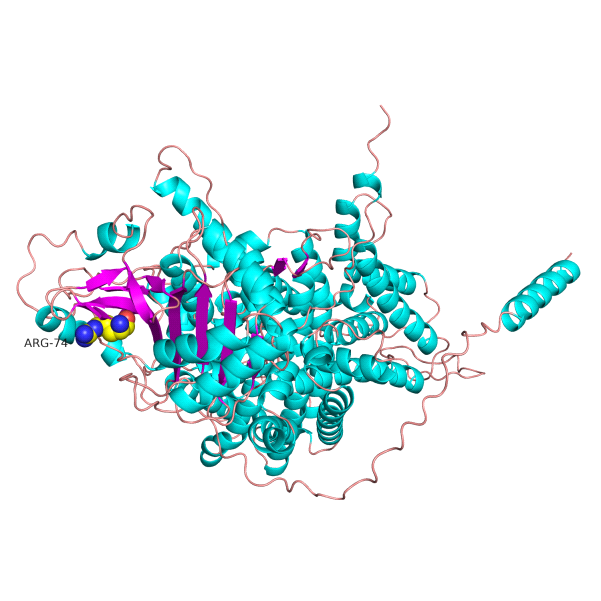
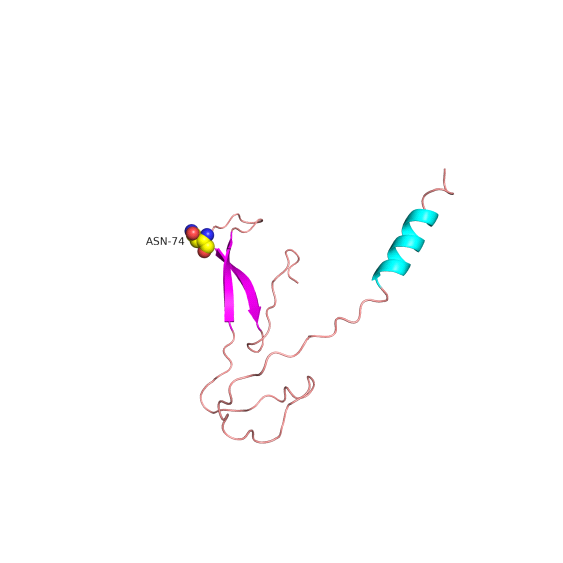
此外,我们通过蛋白质同源建模构建了突变体的三维结构,并与野生型进行比对(图3)。
(A)
(B)
图3 SLC4A1 p.Arg74AsnfsTer35 突变
SLC4A1基因蛋白三维结构对比。(A)野生型蛋白具有完整的全长结构和正确的折叠构象。(B)c.221del移码突变导致蛋白在Asn-74后严重截短,结构完整性丧失,阐释了其致病性的结构基础。
结构模拟显示,SLC4A1 c.221del导致第74位氨基酸由精氨酸(Arg)变为天冬酰胺(Asn)并继续延长35位氨基酸后提前终止,产生严重截短的异常蛋白,其关键功能结构域完全丧失,空间构象发生紊乱。这一结果从分子水平解释了该突变通过破坏红细胞膜骨架蛋白的稳定性,进而引发慢性溶血的分子机制。
同时,家系分析显示患者携带母系遗传的UGT1A1基因顺式杂合变异 c.[-41_-40dup;-3275T>G](图4)。该患者携带UGT1A1单等位基因变异虽不能诊断Gilbert综合征,但多项功能研究已证实其可通过降低基因转录效率,导致UGT1A1酶表达量与功能显著不足,从而使HS疾病状态下的胆红素水平增加,并增加胆石症风险。有研究发现携带UGT1A1*28等位基因数量与HS儿童胆结石发生率之间存在显著关联,具体为(TA)6/(TA)6、(TA)6/(TA)7、(TA)7/(TA)7基因型患者的胆结石发病风险依次增加(图5)[11]。
综上,本研究通过家系分析锁定了分别源于父母双方、影响胆红素代谢不同环节的遗传缺陷,父系的SLC4A1突变通过引发溶血,导致胆红素生成增多;而母系的UGT1A1顺式杂合变异可能进一步升高血清胆红素水平,并增加了胆石症的发生风险。这种生成与清除失衡的协同作用,完整地解释了患者的临床表现。
(A)
(B)
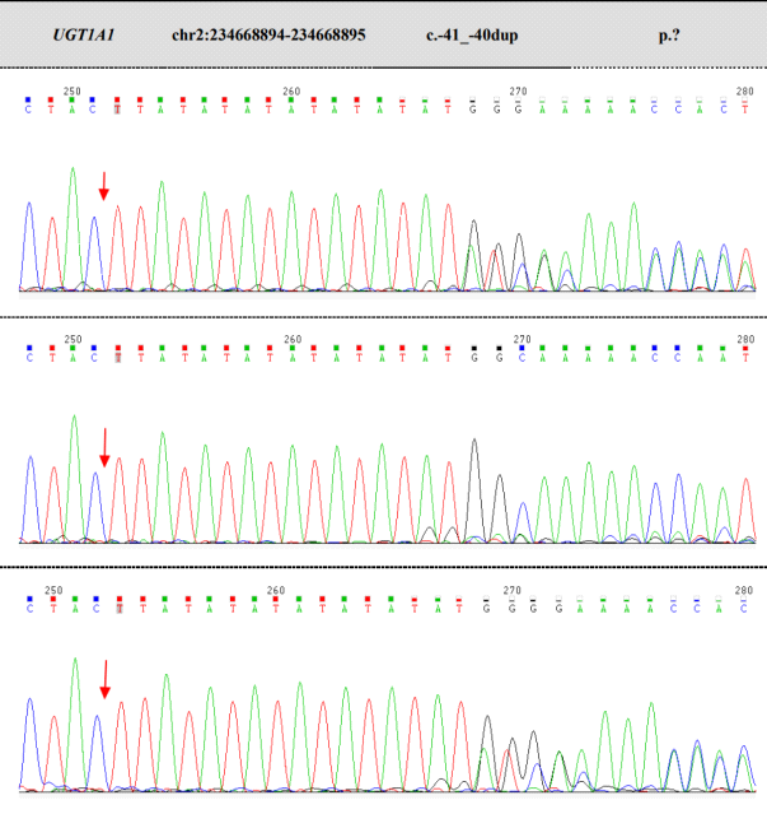
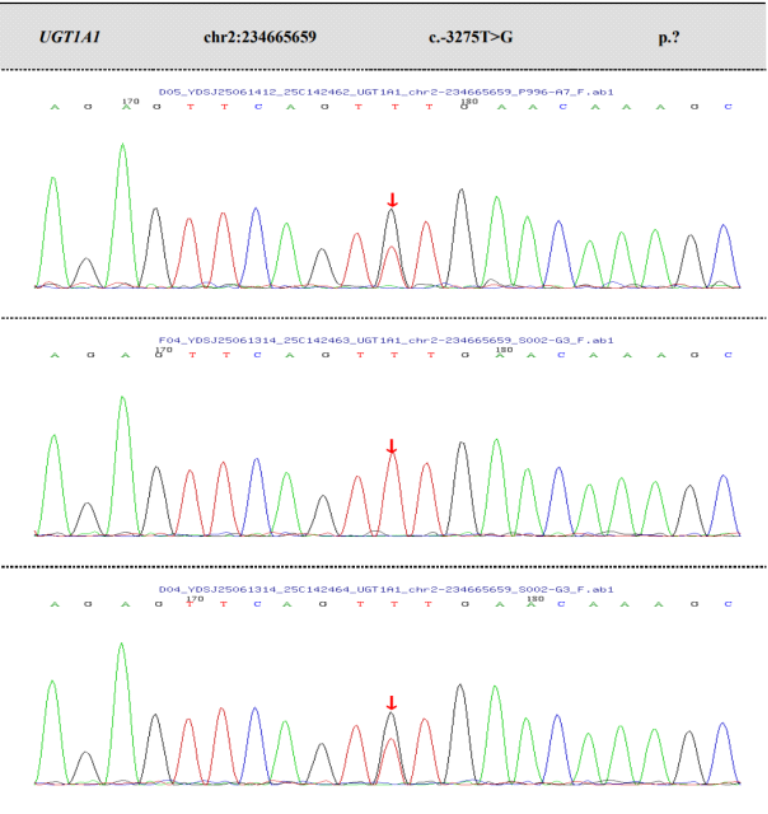
图4 UGT1A1基因启动子区变异(c.-41-40dup与c.-3275T>G)的家系Sanger测序分析
A:测序图谱显示,在UGT1A1基因启动子区c.-41_40dup 位点(箭头所示),在该变异位点下游,先证者(上图)与其母亲(下图)的测序信号表现为持续的序列重叠(套峰),符合TA重复序列杂合插入变异的特征;其父亲(中图)在该位点为野生型单峰。该结果证实此c.-41_40dup 变异来源于母系遗传。
B: 测序图谱显示,在UGT1A1基因启动子区c.-3275T>G 位点(箭头所示),先证者(上图)与其母亲(下图)均在该碱基位置检测到T与G的杂合双峰信号;其父亲(中图)在该位点为野生型(T/T)单峰。该结果证实此c.-3275T>G 点突变来源于母系遗传。
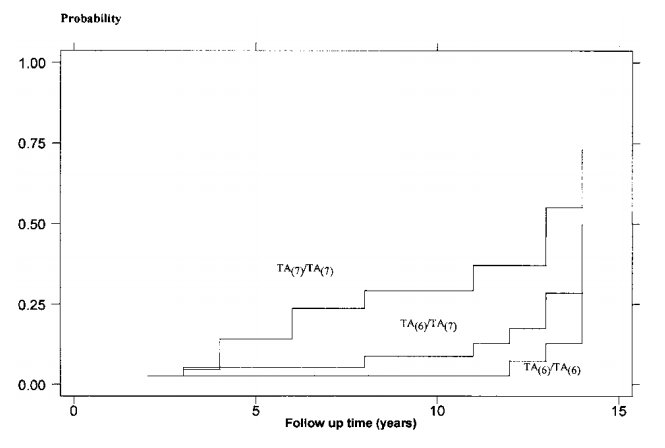
图5 103名遗传性球形红细胞增多症患者形成胆结石的Kaplan-Meier概率,患者根据其UGT1A1基因型进行分类[11]
3.2 供稿专家简介
朱磊
大连医科大学附属第一医院消化内科主任医师、研究生导师
辽宁省基层卫生协会肝病防治专委会副主任委员
辽宁省医学会肝病学会委员
辽宁省免疫学会肝病与免疫分会常委
辽宁省中西医结合学会消化分会委员
大连市中西医结合学会肝病学分会常委、胆胰内镜学会委员

朱连新
大连医科大学附属第一医院,感染科,主治医师
辽宁省中西医结合学会传染病专业委员会委员
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-83997658
▶联系地址:北京市丰台区右安门外西头条8号佑安医院C楼7层