
主办:佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,中华医学会肝病学分会遗传代谢性肝病协作组
总编辑:段钟平
本期责任主编:张岭漪
执行编辑:郑素军,张思琪,侯维
本期目录
一、主编致辞
二、学术进展
三、临床资讯
四、联系方式
月报撰稿及简评专家主要为中华医学会肝病学分会遗传代谢性肝病协作组、佑安肝病感染病专科医疗联盟遗传代谢性肝病专业委员会,以及其他相关领域专家。
“学术进展”:速览最新重要文献,了解遗传代谢性肝病的科研前沿!
“临床资讯”:提供典型病例、名家讲座等信息,共享临床诊治奥妙!
2026年2月28日是第19个国际罕见病日,今年的主题主线仍为“公平(Equity)”,年度口号为“不止罕见(More Than You Can Imagine)”。让我们看见每位患者背后的生活与梦想,推动早诊早治与公平可及的支持,共同点亮希望。
一、主编致辞
遗传性球形红细胞增多症(Hereditary spherocytosis, HS)是一种最常见的遗传性红细胞膜缺陷病,其本质是由于编码红细胞膜骨架蛋白的基因发生突变,导致膜结构稳定性破坏、膜脂质丢失,最终形成球形红细胞[1]。这些变形能力低下的红细胞在脾脏中被过早破坏,引发慢性溶血。该病在北欧裔人群中的患病率约为1/2000,在亚洲人群中也非罕见。约75%的病例为常染色体显性遗传,其余多为常染色体隐性遗传或新发突变[2]。
在分子机制层面,目前已明确的致病基因包括ANK1(锚蛋白-1)、SPTA1/SPTB(α/β-血影蛋白)、SLC4A1(带3蛋白)和EPB42(蛋白4.2)。ANK1蛋白起着核心的“桥梁”作用,连接血影蛋白骨架与脂双层中的带3蛋白。因此,ANK1突变常导致锚蛋白与血影蛋白的联合缺乏。近年来,二代测序技术不断发现新的致病性变异,2025年报道的ANK1基因新发移码突变(p.V1626fs*64)与已知错义突变(p.V463I)以顺式共存,并通过导致突变转录本不稳定而引起疾病[3]。同时,SPTA1基因的无义突变若与α-血影蛋白LELY这类低表达等位基因形成复合杂合状态,则可能导致常染色体隐性遗传的严重表[4]。
HS的临床表现谱系宽广,从无症状携带者到需定期输血的严重贫血患者均存在。典型特征包括慢性溶血症候群:贫血、间歇性黄疸、脾肿大和色素性胆结石。诊断流程始于临床怀疑,血涂片发现球形红细胞、网织红细胞计数增高和Coombs试验阴性是初始线索。传统实验室诊断的“金标准”是孵育后红细胞渗透脆性试验,而流式细胞术嗜酸性-5-马来酰亚胺结合试验因其快速、标准化已成为重要的筛查工具。值得注意的是,EMA结合试验在某些特定基因型(如部分SPTA1突变)中可能显示正常结果,因此阴性不能完全排除HS,此时基因测序成为确诊和分型的关键[5]。
治疗决策需高度个体化。所有患者建议长期补充叶酸。脾切除术是唯一能显著改善溶血和贫血的治疗手段,但因术后存在凶险性感染和远期血栓风险,手术时机需慎重权衡,通常建议在6岁后进行。对于重症幼儿,部分脾切除术作为一种过渡性治疗,可在缓解溶血的同时保留部分免疫功能。胆石症是常见并发症,有症状者需行胆囊切除术。
肝脏异常是HS管理中的关键环节,主要由三方面因素导致:首先,慢性溶血使肝脏持续承受非结合胆红素转化的负荷,导致间歇性非结合型高胆红素血症。其次,胆色素结石发生率显著增高,尤其是在合并Gilbert综合征(UGT1A128等位基因纯合子)的患者中,风险可增加4~5倍,导致的胆道梗阻可引起结合胆红素升高和碱性磷酸酶升高,即继发性肝损伤。最后,长期慢性溶血及反复输血可能导致继发性血色病,引起肝铁过载,长期可致肝纤维化甚至肝硬化。因此,对HS患者的长期管理必须包括定期监测肝功能、腹部超声评估胆道系统以及血清铁蛋白水平,以实现对肝脏并发症的早期发现和干预[6]。
HS的管理已进入一个融合了经典血液病学、精准分子诊断和长期多器官随访的综合时代。对基因型-表型关系的深入理解,有助于实现更个体化的风险评估和治疗选择。
本期月报报道1例遗传性球形红细胞增多症引起的高胆红素血症患者,患者胆红素升高以间接胆红素升高为主,血常规血红蛋白正常,网织红细胞百分率升高为14.7%,肝脏穿刺病理提示:汇管区纤维组织增生,以淋巴细胞,浆细胞为主的炎性细胞浸润,胆管结构基本正常,呈轻度界面炎病理改变。此后依据临床表现及相关检验报告,进一步完善全外显子基因测序,确诊SPTB基因缺失突变导致的遗传性球形红细胞增多症。患者母亲与姐姐也有间断胆红素升高,后经家系验证确定该突变遗传来自母亲,遗传方式为常染色体显性遗传。常染色体显性遗传是HS主要的遗传方式,其子女有50%概率遗传该突变,后期也需要进一步对患者姐姐进一步验证。希望通过分享此例病例,加深临床医师对遗传性球形红细胞增多症相关高胆红素血症的认识,拓展黄疸患者,以间接胆红素升高为主肝功能异常患者的鉴别诊断思路。
参考文献
[1] Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411-1426.
[2] Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21(1):1-20.
[3] Bogusławska DM, Rybka J, Koszela P, et al. Two Variants of the ANK1 Gene Associated with Hereditary Spherocytosis. Biomedicines. 2025;13(2):308.
[4] Molina-Arrebola MA, Bain BJ. Hereditary Spherocytosis due to an SPTA1 Nonsense Mutation Coinherited With α spectrinLELY in Trans. Am J Hematol. 2025;100(12):2355-2356.
[5] Bolton-Maggs PH, Langer JC, Iolascon A, et al. General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012;156(1):37-49.
[6] del Giudice EM, Perrotta S, Nobili B, et al. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood. 1999;94(7):2259-2262.

张岭漪
兰州大学第二医院 肝病科 教授 主任医师 研究生导师
中华医学会预防感染病防控分会 委员
中国医疗保健国际交流促进会 肝胆疾病学分会 常务委员
中华医学会肝病学分会肝纤维化学组委员、终末肝学组委员
二、学术进展
Pubmed最新重要文献速览(长按文末二维码或“阅读原文”可下载)
1.先天性红细胞疾病患者胆结石的10年风险:一项全国性队列研究(American Journal of Hematology,2025, IF=10.3; Q1区)

2.一例年轻女性梗阻性黄疸发作背后的隐匿病因(Gastroenterology,2026, IF=25.9; Q1区)
3.遗传性球形红细胞增多症:离子转运缺陷与渗透梯度变形指数测定(OGE)谱的关联——综述(International Journal of Molecular Sciences,2026, IF=4.9; Q1区)
4.当一种特征演变为疾病:镰状细胞携带者状态与遗传性球形红细胞增多症罕见的血液学重叠病例(American Journal of Hematology,2026, IF=10.3; Q1区)
5.遗传性球形红细胞增多症中的铁过载:遗传因素是否为病因?(British Journal of Haematology,2026, IF=3.9; Q1区)
6.遗传性球形红细胞增多症中的认知障碍(British Journal of Haematology,2026, IF=3.9; Q1区)
7.由SPTA 1无义突变与α spectrinLELY共同遗传导致的遗传性球形红细胞增多症(American Journal of Hematology,2025, IF=10.3; Q1区)
8.肝门部肝黏液性囊性肿瘤致梗阻性黄疸:罕见病例报道及文献综述(Current Oncology,2025, IF=3.4; Q2区)
9.伴十二指肠–胆道瘘的梗阻性黄疸:诊断与治疗挑战——病例报道及文献综述(Frontiers in Medicine,2025, IF=3.0; Q1区)
10.遗传性球形红细胞增多症中的脑血管受累:观察性队列研究与病例-对照MRI研究(Orphanet Journal of Rare Diseases,2025, IF=3.2; Q2区)
三、临床资讯
3.1 病例分享:1例遗传性球形红细胞增多症黄疸患者
患者男性,17岁,因“发现皮肤黏膜及巩膜黄染2年余”入院。
现病史:患者2年前因发现皮肤黏膜及巩膜黄染,就诊于当地医院查肝功能:总胆红素TBIL 219.0μmol/L,直接胆红素DBIL 17.2μmol/L,间接胆红素IBIL 201.8μmol/L,转氨酶正常。病毒性肝炎标志物、ANA、AMA-M2、抗GP210等免疫指标未见异常,B超提示门静脉增宽,脾大(轻度)。平素无乏力,无厌油、纳差、皮肤瘙痒等不适症状。此后患者间断复查肝功能,胆红素升高,以间接胆红素升高为主,总胆红素波动于70.3-219.0μmol/L之间,间接胆红素波动在55.7-201.8μmol/L。 ALP、GGT时有升高(ALP 135.9-256.8 U/L, GGT 34-298.6 U/L、ALT 24-119 U/L、AST 17- 97 U/L)。间断口服熊去氧胆酸、复方甘草酸苷保肝治疗后胆红素可短暂下降。为进一步诊治就诊于我院门诊。患者平素体健,精神、食欲、睡眠可,大小便正常,体重稳定。
既往史、个人史、家族史:母亲、姐姐发现胆红素升高。否认服药史,无饮酒史。父亲正常。
查体:体温36.5℃,血压120/89mmHg,身高3750px,体重60kg,BMI 26.7kg/m2,神志清楚,营养良好,皮肤巩膜黄染,心肺查体未见异常,腹软,肝脾未触及,双下肢无水肿。
实验室检查:
肝功能:
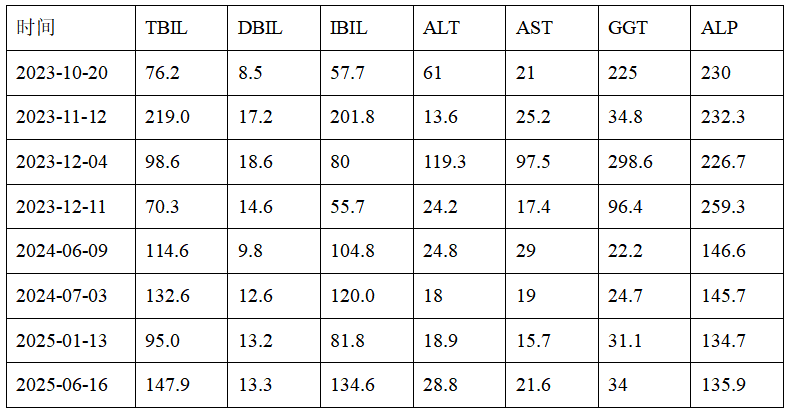
图1.患者肝功能变化
一般检查:血常规、凝血、甲状腺功能、甲胎蛋白正常;
肝脏病因检查:
HBV(-),HCV(-),CMV(-),EBV(-),HIV(-),ANA(-),AMA-M2(-),gp210(-),sp100(-),ANCA(-);免疫球蛋白、铜蓝蛋白正常;coombs实验阴性。外周血细胞分类:成熟粒细胞流式细胞免疫表型检查:阴性。红细胞寿命呼气实验检验提示:红细胞平均寿命只有 14 天(正常大约在120天左右),提示严重溶血可能。血红蛋白(Hb)>120 g/L,提示目前溶血处于代偿期。血浆游离血红蛋白(FHB)191.67 mg/L(参考值<50mg/L)。
影像学检查:
腹部超声:脾大,脂肪肝。腹部CT:脂肪肝,脾大,腹膜后肠系膜区增大淋巴结。
肝穿病理:
描述穿刺组织肝小叶结构较完整,汇管区纤维组织增生,以淋巴细胞,浆细胞为主的炎性细胞浸润,胆管结构基本正常,呈轻度界面炎病理改变,小叶内可见多灶、碎片状坏死,于中央静脉周围可见碎片状坏死,部分肝细胞胞浆疏松,淡染,呈水肿表现。(肝脏):形态学符合慢性肝炎(G2S2)。该病变模式常见于病毒性肝炎,自身免疫性肝炎,DILI等,请结合临床相关检查。免疫组化:A组织块:CK7(胆管+);CD10(胆小管+)。特染:A组织块:Masson(网状支架+,汇管区纤维组织轻度增生);D-PAS(-);普鲁士蓝(局灶+);刚果红(-);网状纤维(网状支架+,汇管区纤维组织轻度增生)。
全外显子基因测序:
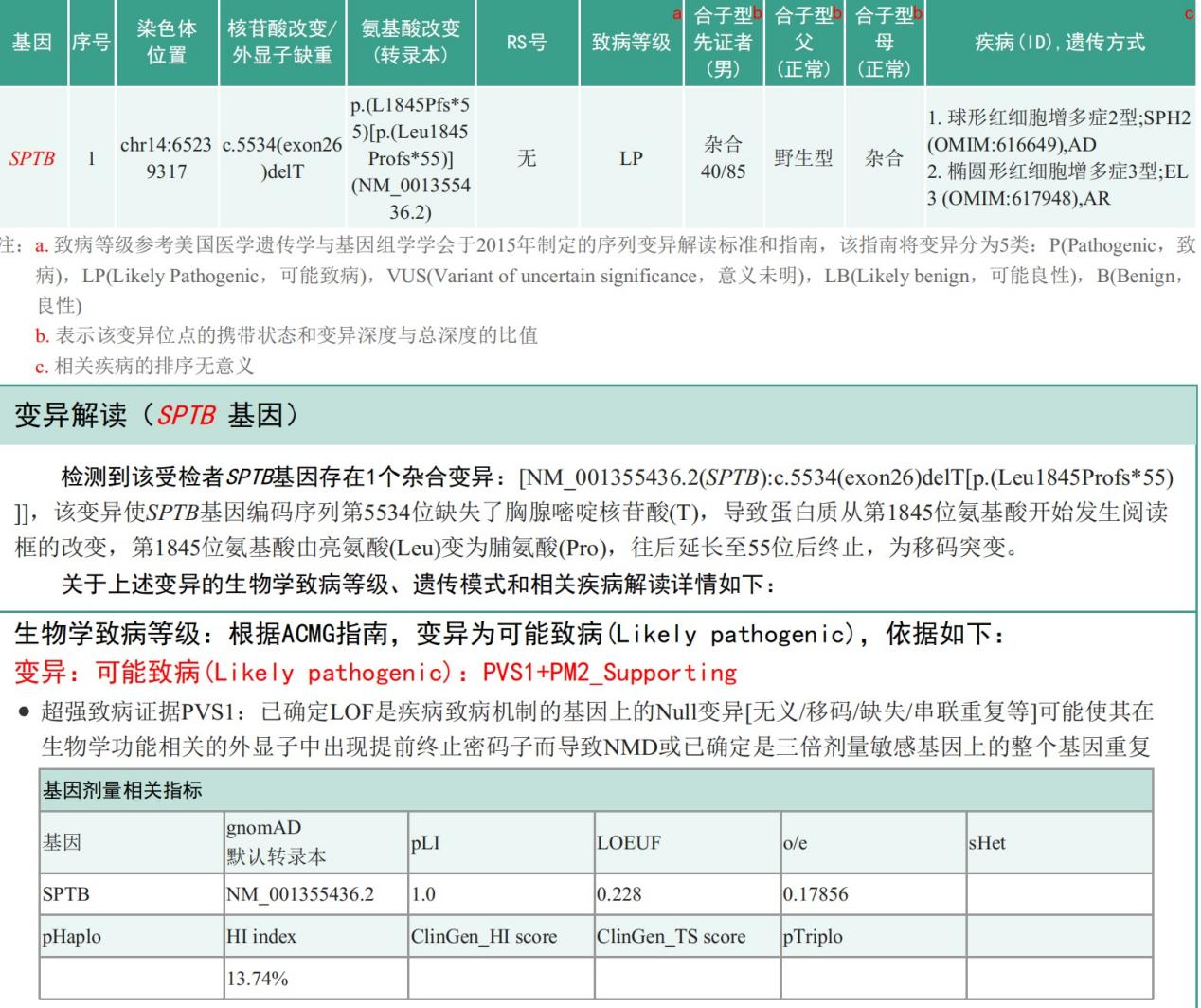
图2:致病可能性较高的基因变异SNV检测结果
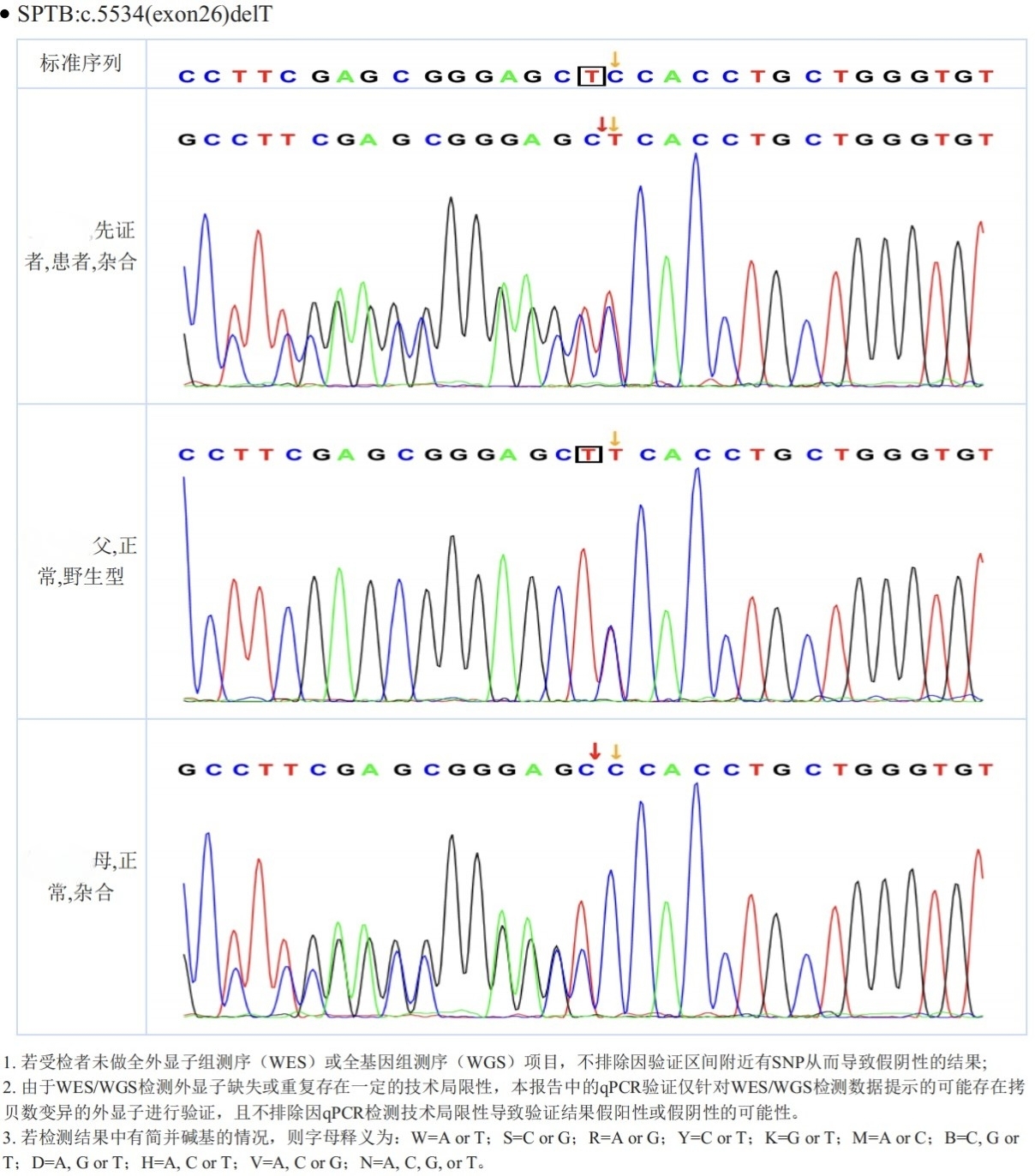
图3:致病可能性较高基因变异家系验证结果
从上图我们注意到,患者及其父亲还存在SPTB c.5535 C>T,该位点为在ClinVar数据库中标记的为良性变异,p.Leu1845=(同义变异),也就是不改变氨基酸。
病例特点:1.青少年男性,慢性病程。2.肝功能异常表现为胆红素升高,间接胆红素升高为主。3.母亲,姐姐均亦发现胆红素升高。4.基因检测提示SPTB基因缺失突变,家系验证确定该突变遗传自母亲。
诊断:遗传性球形红细胞增多症,脾大,脂肪肝。
治疗:建议行脾脏切除治疗。患者由于个人原因暂为进行脾切手术,由于患者目前未出现胆石症情况,可择期再行脾切手术,后续仍需继续随访病情进展情况。
3.2 供稿专家简介
张岭漪
兰州大学第二医院 肝病科 教授 主任医师 研究生导师
中华医学会预防感染病防控分会委员
中国医疗保健国际交流促进会肝胆疾病学分会常务委员
中华医学会肝病学分会肝纤维化学组委员、终末肝学组委员

裴志燕
兰州大学第二医院,肝病科,主治医师
四、联系方式
▶投稿邮箱:yichuandaixie2020@163.com(用于征集典型病例、PPT或委员会动态信息)
▶联系电话:010-83997658
▶联系地址:北京市丰台区右安门外西头条8号佑安医院C楼7层